The Complement System
Research Goal
In our research, we aim to resolve the molecular mechanisms underlying the complement pathways.
- Enhancing complement activity by properdin
- Formation of IgM-C1-C4b complexes
Recently added topics:
The complement system is an evolutionary ancient immune defense mechanism, present in vertebrates and invertebrates. It forms a first line of defense in plasma and fluids surrounding tissues and enables the host to recognize and clear invading pathogens (bacteria and viruses) and altered host cells while protecting healthy host cells and tissues. In humans this system is formed by more than 30 large modular plasma proteins and cell surface receptors.
Classical pathway:
In the classical pathway of complement activation the C1 complex binds and is activated by e.g. antibodies bound to antigens on a surface. We resolved the quaternary arrangements of IgG and IgM antibodies that bind and activate C1.
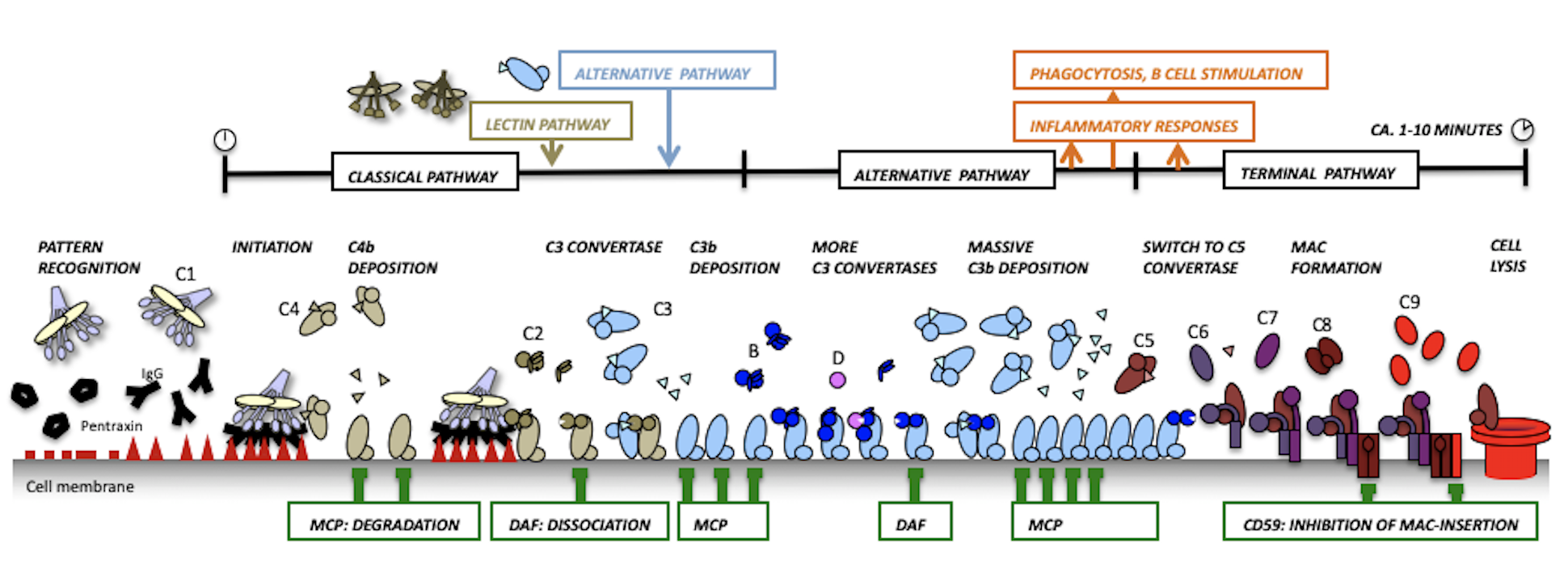
Alternative pathway:
We have resolved the structural details and elucidated the molecular mechanisms of the central components of the complement system that lead to amplification of the complement response and is referred to as the Alternative Pathway of complement activation. In short, we revealed that upon proteolysis and complex formation large domain-domain rearrangements of these multi-domain proteins occur that determine the specificity needed for a local and brief burst of activity marking targets cells for immune clearance.
- Activation states of C3
- Serine protease factor B
- Convertase formation
- C3 convertase activity and specificity
- Enhancing complement activity by properdin
Topics:
Host protection:
To protect healthy host cells from complement attack, the host has a range of complement "regulator" molecules. We determined structures of the abundant soluble human regulator (factor H), human membrane bound (DAF, MCP and CR1) and a homologue from small-pox virusse (SPICE), bound to the central activated molecule C3b that blocks complement amplification. Furthermore, we resolved the tri-partite structure of C3b-Factor H-Factor I, which revealed the molecular mechanisms underying cofactor activity.
Terminal pathway:
One of the effector functions of complement activation is formation of the membrane-attack complex (MAC) that forms 100 Å pores in membranes. Structural details of the central MAC/perforin (MACPF) domain and the structure of the first complex C5b6 provided insights into MAC initiation and formation.
- Membrane-attack-complex
Topics:
Surface-bound immune complexes invoke complement activation through binding and activating the C1 complex of the complement system. Initiation of the proteolytic cascade of complement through C1 is commonly referred to as the “classical pathway” of complement activation.
Antibody-mediated complement activation may induce cell lysis (referred to as complement-dependent cytotoxicity (CDC) of an antibody, or lead to ‘silent’ (non-inflammatory) of apoptotic host cells. The presence of allo-antibodies (host antibodies against donor antigens) induces antibody-mediated rejection (AMR) of transplanted organs. In contrast, directing antibodies against tumour markers is beneficial in reducing tumours. By studying the molecular processes of complex formation, we hope to address questions, such as what determines the CDC of an antibody and how does IgM stimulate ‘silent’ removal of apoptotic cells?
IgG antibodies form Fc hexamers on cells
In collaboration with Paul Parren (Genmab, Utrecht) and Bram Koster (EM goup at LUMC, Leiden), we revealed the arrangement of IgG1 antibodies needed on liposomes or cells that binds and activates C1 (Diebolder, Beurskens et al. Science 2014).
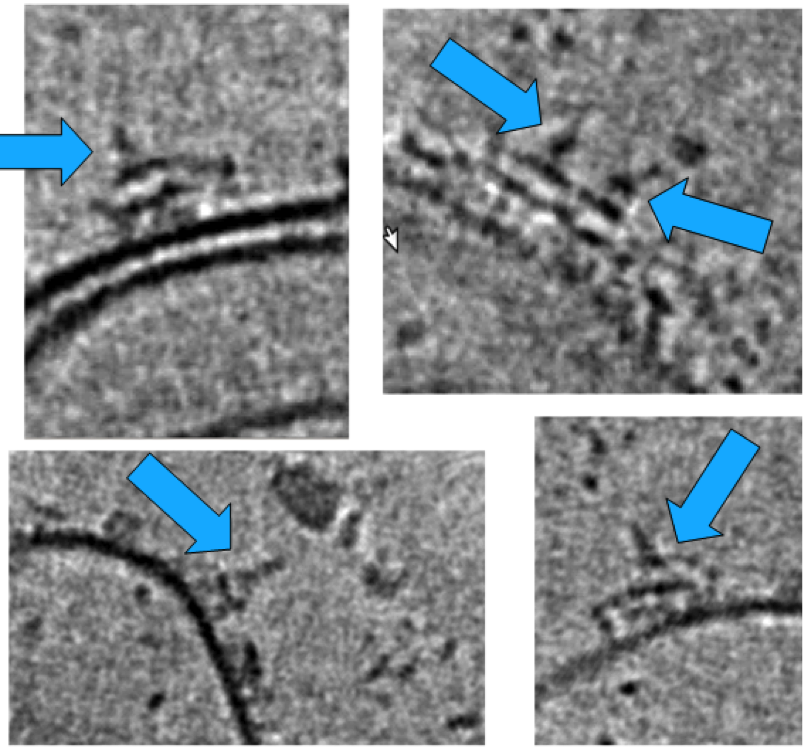
We used a liposomal model system consisting of PC/PE lipids with dinitrophenyl (DNP) haptens on the surface (see Yamamoto et al.) for testing complement lysis activity and imaging by cryo-electron tomography (cryo-ET). Addition of anti-DNP polyclonal antibodies yielded patches of single layers of antibodies on the vesicle surface (left figure). C1 bound on top of these antibody structures (blue arrows in right figure). Over 200 tilt series of these Ab-C1 complexes were collected using a Krios Titan at NeCEN. Class averaging selected ~100 complexes resulting in an electron density map at ~6.5-nm resolution. The resulting density map was consistent with six antibodies forming a Fc-hexamer ring with for each IgG one Fab in the plane and one Fab bound to the surface. The C1q headpieces were bound to the periphery of the Fc hexamers ring. Specific mutations at the Fc-Fc interface enhanced antibody-mediated complement activation against tumour models in vitro. Modifying monospecific (bivalent), non-complement activating, IgG antibodies into bispecific antibodies turned on complement activation. This switch strongly supports the structural data that complement activation by antibodies requires binding of one Fab arm to the target and one Fab "free", which allows formation IgG hexameric Fc platforms.
Binding and activation of C1 to hexameric IgG1-Fc platforms
Next, we used triple ‘RGY’ mutants of IgG1, which form stable hexameric complement-activating Fc platforms in solution for cryo-EM single particle analysis to obtain higher resolution structural insights of C1-IgG16 complexes. The data revealed a dynamical arrangement of C1q binding to the Fc platforms yielding medium resolution density maps at ca. 10-Å resolution for the whole C1-IgG16 complex and 7-Å resolution for the hexameric Fc platform that bindig the globular domains of C1q [figure or movie](Ugurlar et al. Science, accepted). The data revealed that C1q binds Fc in an ‘open’ CH2-CH2’ configuration yielding two distinct binding site on CH2 (FG loop) and its partner CH2’ (BC and DE loops) Fc domains. Furthermore, the data strongly suggests that the C1 collagen arms are pulled together upon binding IgG1-Fc platforms. We speculated that this compaction represents a conformational trigger for C1 activation upon surface binding. Alternatively, liposomes with densely packed IgG1-C1 complexes would suggest potential cross-activation between complexes, similar to what is proposed by Steffen Thiel for activation of MBL-MASP22 complexes by MBL-MASP12 complexes (Kjaer et al. and Thiel, Structure 2015). In contrast to MASP complexes, C1 carries both two molecules C1s (structurally and functionally homologous to MASP2) and two molecules of C1r required for activation of C1s. When and how either MASP1 or C1r become activated remains unclear.
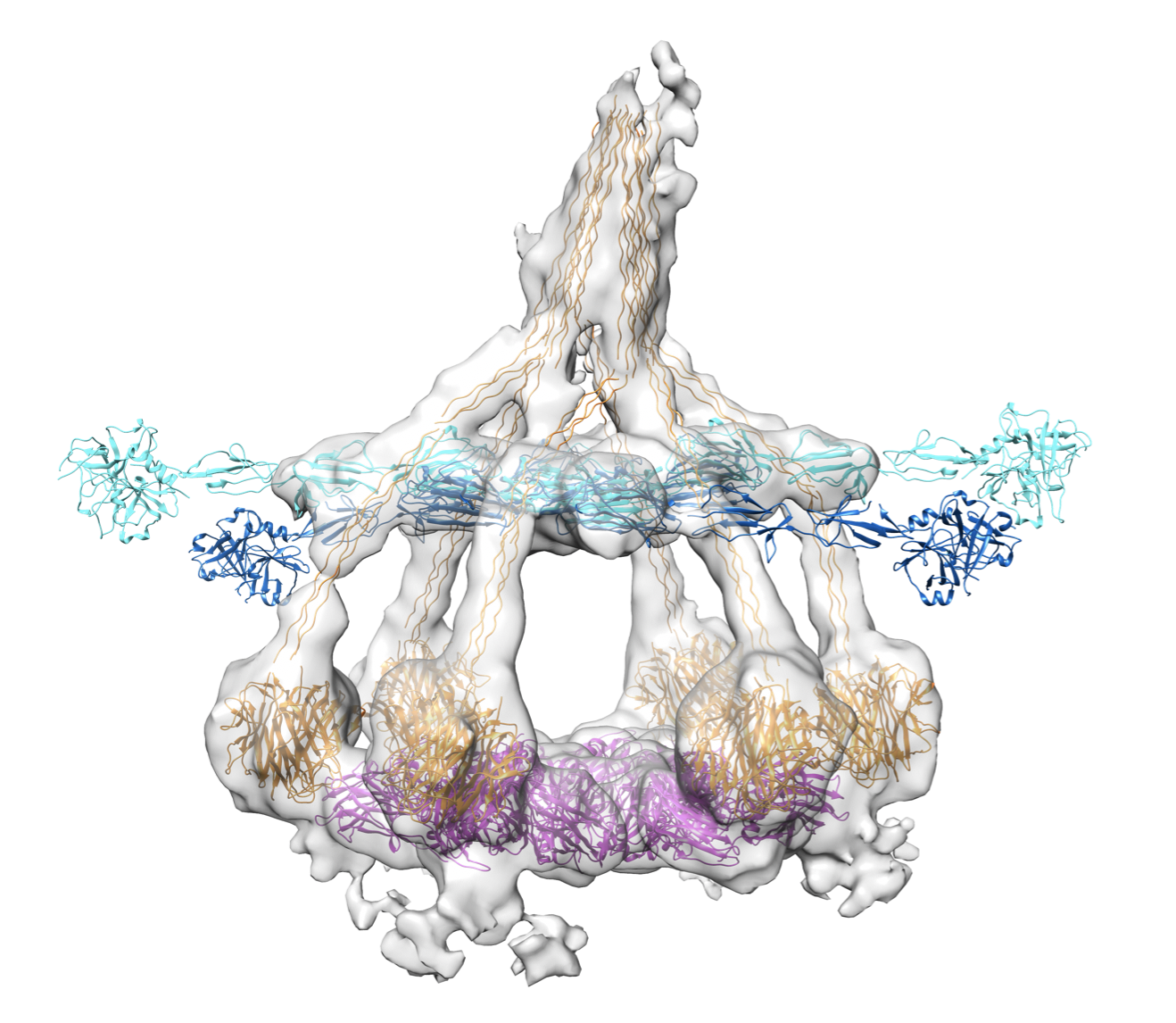
We have imaged the first steps of IgM-mediated complement activation on liposomes by cryo-EM tomography, by mixing ca. 100-200 nm sized liposomes bearing CD52 epitopes with anti-CD52 IgM and human serum at low temperature, i.e. 4℃ (Sharp et al. PNAS 2019).
Cryo-EM tomography resolved structures of pentameric and hexameric IgMs in complex with C1 (C1qr2s2) bound to the liposomes. To our surprise, these complexes also contained C4b, indicating that complement is stopped at 4℃ at the step of C4b release and C2 binding in the formation of 'classical pathway' C3 convertases (C4b2a).
The structures of both pentameric and hexameric IgM adopted hexagonal arrangements of their Cμ3-4/3'-4' domains to bind C1q of the C1 complex, with their dimeric hinge domains Cμ2/2' bending downwards connecting to both Fab arms that were bound to surface antigens. The hexagonal arrangements were similar to that of the Cγ2-3 domains in the Fc platform of hexamerized IgG1 molecules (Ugurlar et al., Science 2018; Diebolder et al. Science 2014). C1q globular domains bind these IgM platforms differently than the IgG1-Fc platforms yielding a much wider spreadig of the six C1q-collagen helices, compared to C1q-collagen helices bound to hexamerized IgG1. Densities for the C1-associated proteases C1r and C1s was present for all six domains (CUB1-EGF-CUB2-CCP1-CCP2-SP) of the four molecules, with C1r and C1s forming a hetero-tetramer of CUB1-EGF-CUB2 domains in between the collagen legs of C1q and CCP1-CCP2-SP of C1r pointing outwards and CCP1-CCP2-SP of C1s bending down with the catalytic SP domains of C1s contacting the (one or two) bound C4b molecules.
We determined three-dimensional structures of the central complement component C3 in three activation states, native C3, activated C3b and inactive C3c.
C3 is a large (1,641 amino-acid residues) protein with a remarkable structural arrangement consisting of 13 domains formed by two protein chains β and α (Janssen et al. Nature 2005). The internal domain homologies indicate that this type of protein molecule evolved from a core of eight homologous "macroglobulin" (MG) domains, marking the beginning of a generic host defense mechanism (more than 1,300 million years ago; well before the emergence of antibodies). C3 is critical to label cells and cellular debris for immune clearance. This labelling is achieved through covalent attachment of a reactive thioester. In native C3 the thioester is protected in two ways. First, the thioester is sequestered from water to prevent hydrolysis and second, domain-domain interactions prevent transformation of the reactive thioester into a highly reactive thiolate and acyl-imidazole intermediate.
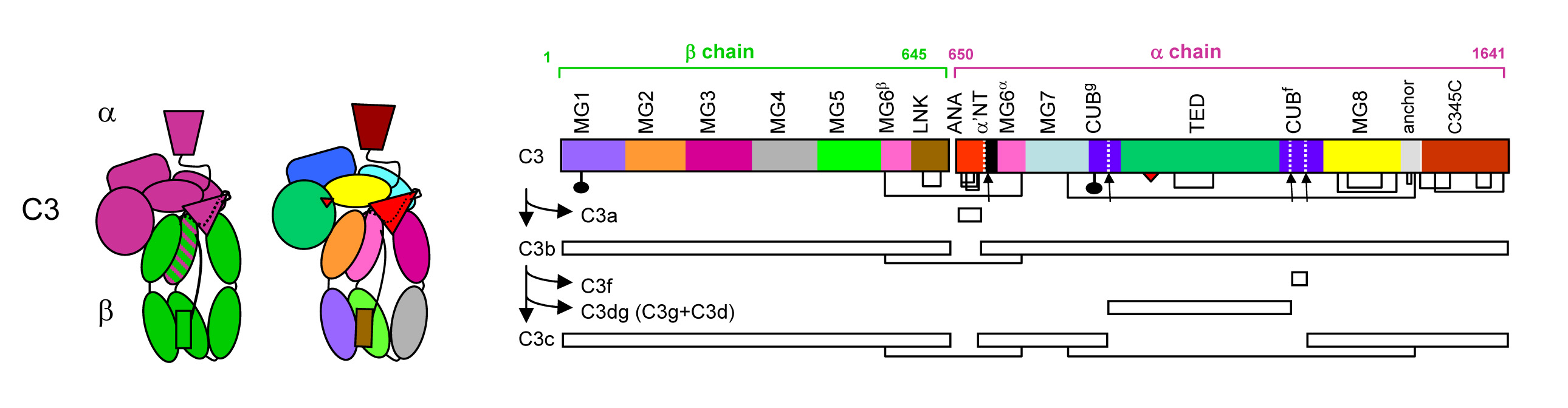
Proteolytic activation of C3 produces C3b. The structure of C3b changes markedly compared to that of native C3 (Janssen et al. Nature 2006). The thioester region moves over 85 Å, becomes fully exposed and is activated into the acyl- imidazole intermediate for reaction with hydroxyls present on the targeted surfaces resulting in covalent attachment. Moreover, the rearranged structure now exposes previously hidden binding sites for pro-enzyme factor B and a multitude of regulator proteins (for decay accelerating activity and cofactor activity).
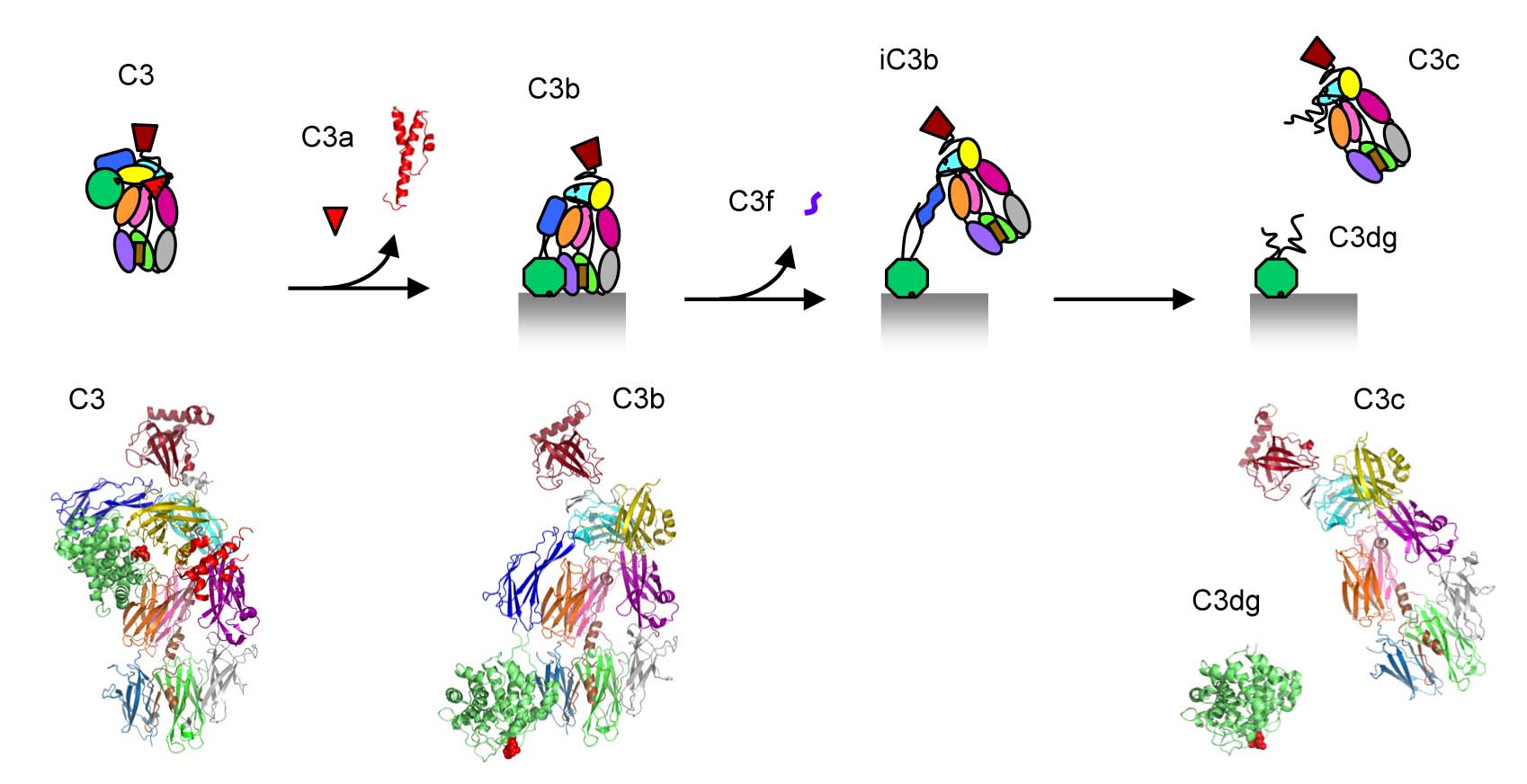
Binding of factor B to C3b is the first step in an assembly process that yields the protease complex C3bBb, which is the central C3 convertase (of the so- called alternative pathway). This C3 convertase cleaves more C3 into C3a and C3b, hence amplifies the complement response yielding massive C3b labelling of the target surface. Surfaces covered by C3b, and the subsequent proteolytic fragments iC3b and C3dg, are marked for destruction and clearance; either through initiation of the terminal pathway of complement leading to cell lysis, binding to macrophages for phagocytosis or binding to B-cells stimulating the adaptive immune response.
Factor B is the pro-protease that after assembly and proteolysis yields the central protease complex C3bBb responsible for amplification of the complement response through the so-called alternative pathway of complement activation.
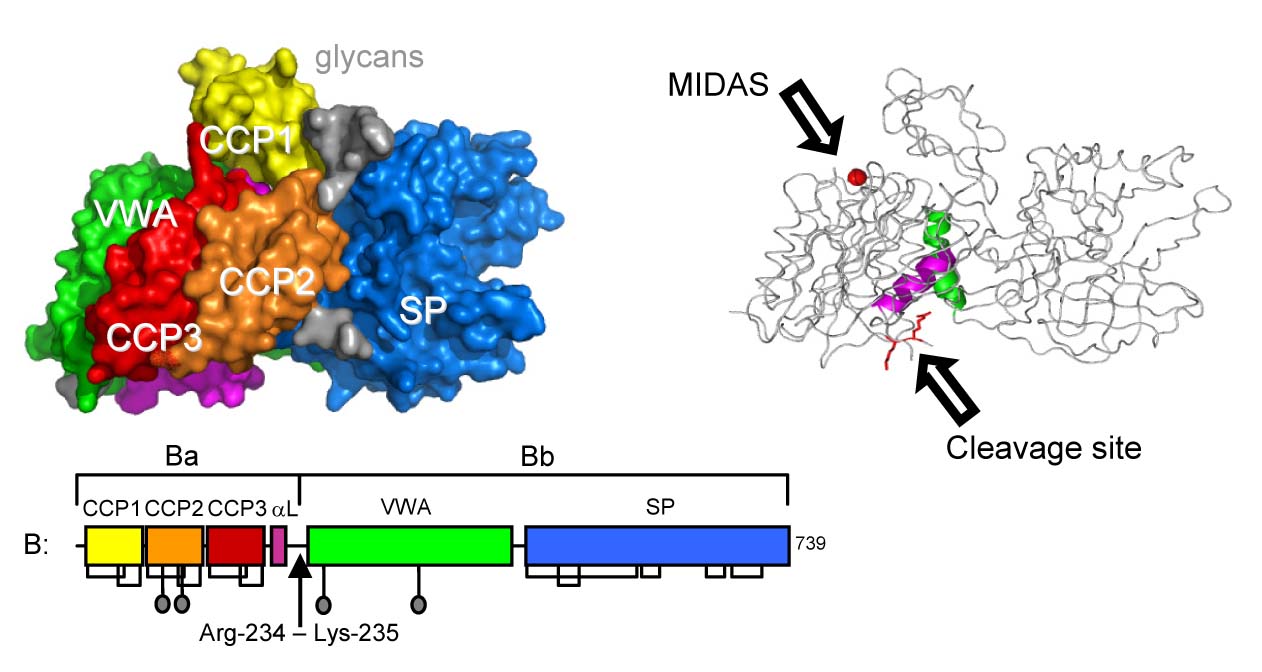
We determined the structure of full-length pro-protease factor B (Milder et al. Nature SMB 2007) that consists of three N-terminal complement-control-protein (CCP) domains and a linker helix αL (forming the pro-peptide segment Ba), and a central Von Willebrand A-type (VWA) domain and C-terminal serine protease (SP) domain (together forming the protease segment Bb). The VWA domain is homologous to the regulatory "inserted" (I) domains of integrins. Similar to integrin I-domains, VWA has a metal-ion (Mg2+) dependent adhesion site (MIDAS) critical for ligand (C3b) binding and a C-terminal helix α7 that may be involved in determining ligand-binding affinity at the MIDAS and transmitting conformational changes upon ligand binding (Milder et al. Structure 2006).
In the pro-enzyme factor B, however, we observe a novel arrangement of these elements in the VWA domain. Helix α7 is displaced from its canonical groove by helix αL of the pro-peptide and the MIDAS, which is critical for C3b binding, is abrogated. Furthermore, the P1 arginine residue of the scissile bond cleaved by factor D in activation of the pro-convertase C3bB is located in a shallow cleft formed by the helices αL and α7 and forms salt bridges with both helices. Thus, these arrangements suggest a "locked" state for native factor B and that binding of C3b likely induces large conformational rearrangements putatively of the helices αL and α7 that expose the scissile bond and make factor B susceptible to cleavage by factor D.
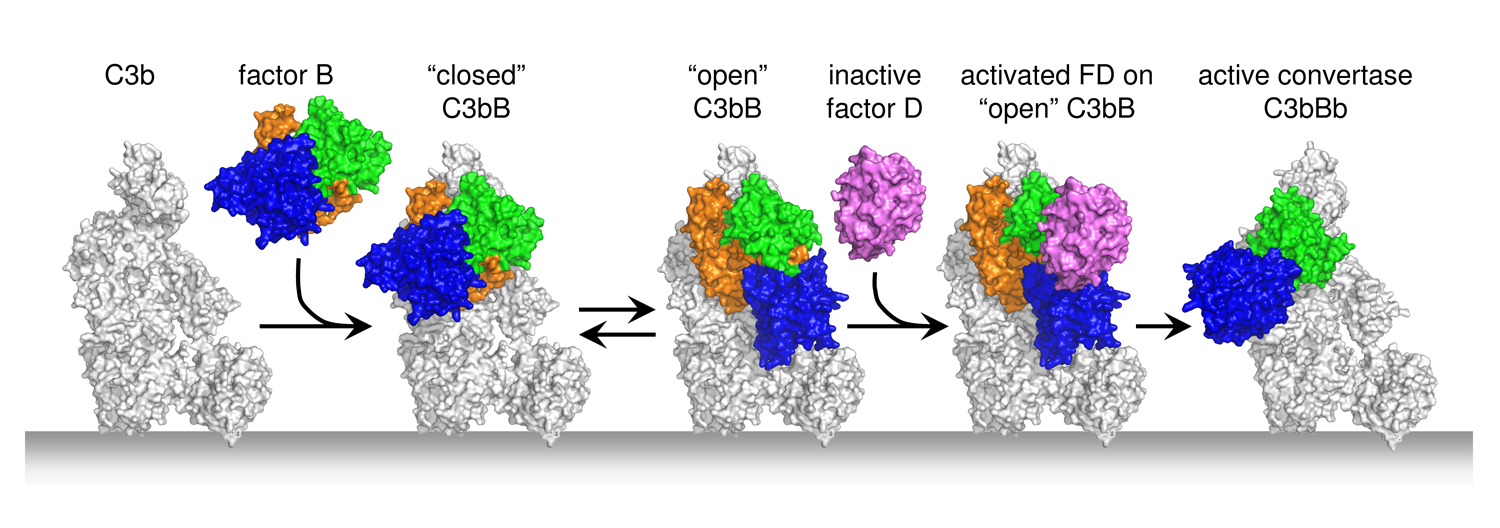
The central C3 convertase of the alternative pathway, that is responsible for the amplification of the complement response, is formed by surface-bound C3b and pro-protease factor B in a two step assembly process. First, factor B binds in an Mg2+-dependent manner to C3b yielding the pro-convertase C3bB. Second, factor D cleaves C3bB, which releases the pro-peptide fragment Ba resulting in the active C3 convertase of the alternative pathway C3bBb.
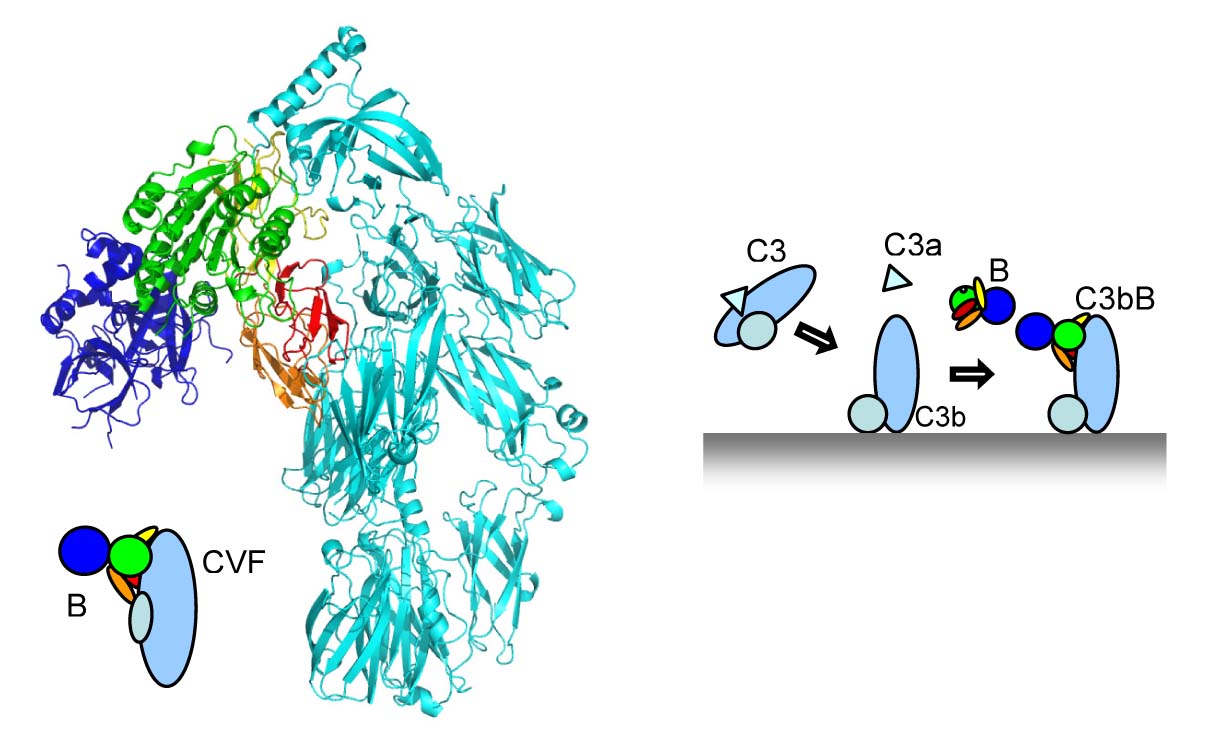
We studied the pro-convertase formed by cobra-venom factor (CVF, a potent homologue of C3b) and factor B (Janssen et al. EMBOJ 2009). The structure revealed that CVF is structurally homologous to C3b. Factor B binds CVF through two interfaces, one formed by the CCP domains of the pro-peptide segment Ba and one through the VWA domain of the protease segment Bb. The interactions with the Ba segment are apparently critical to "load" factor B onto CVF or C3b, because the fragment Bb alone does not bind CVF or C3b. A critical interaction is provided by the metal-ion (Mg2+) adhesion site (MIDAS) present in the VWA domain of factor B, where the C-terminus of CVF chelates the Mg2+ ion. Surprisingly, we observe no overall domain rearrangement in factor B upon binding to CVF. Notably, the scissile bond remains occluded as in the structure of native factor B. EM data from the Llorca lab (Madrid) indicates, however, conformational changes as factor B binds to C3b. We hypothesize that CFV-B may represent a "loading" state whereas the EM data of C3bB represents an "activation" state of the pro-convertase, which can be cleaved by factor D.
The mammalian complement system clears invading pathogens and cellular debris, such as apoptotic host cells, from blood and fluids surrounding tissues. All complement activation routes converge into the amplification loop of the so-called "Alternative Pathway", where C3 is activated by C3 convertases into anaphylatoxin C3a and opsonin C3b. These C3 convertases are surface-bound, short-lived protease complexes generated by C3b, pro-protease factor B and protease factor D. We have resolved the structural details of complex formation of the pro-convertase formed by C3b and pro-enzyme factor B (C3bB) and the activation of C3bB by the protease factor D (C3bBD) that yields the active C3 convertase (C3bBb) of the Alternative Pathway (Forneris et al. Science 2010).
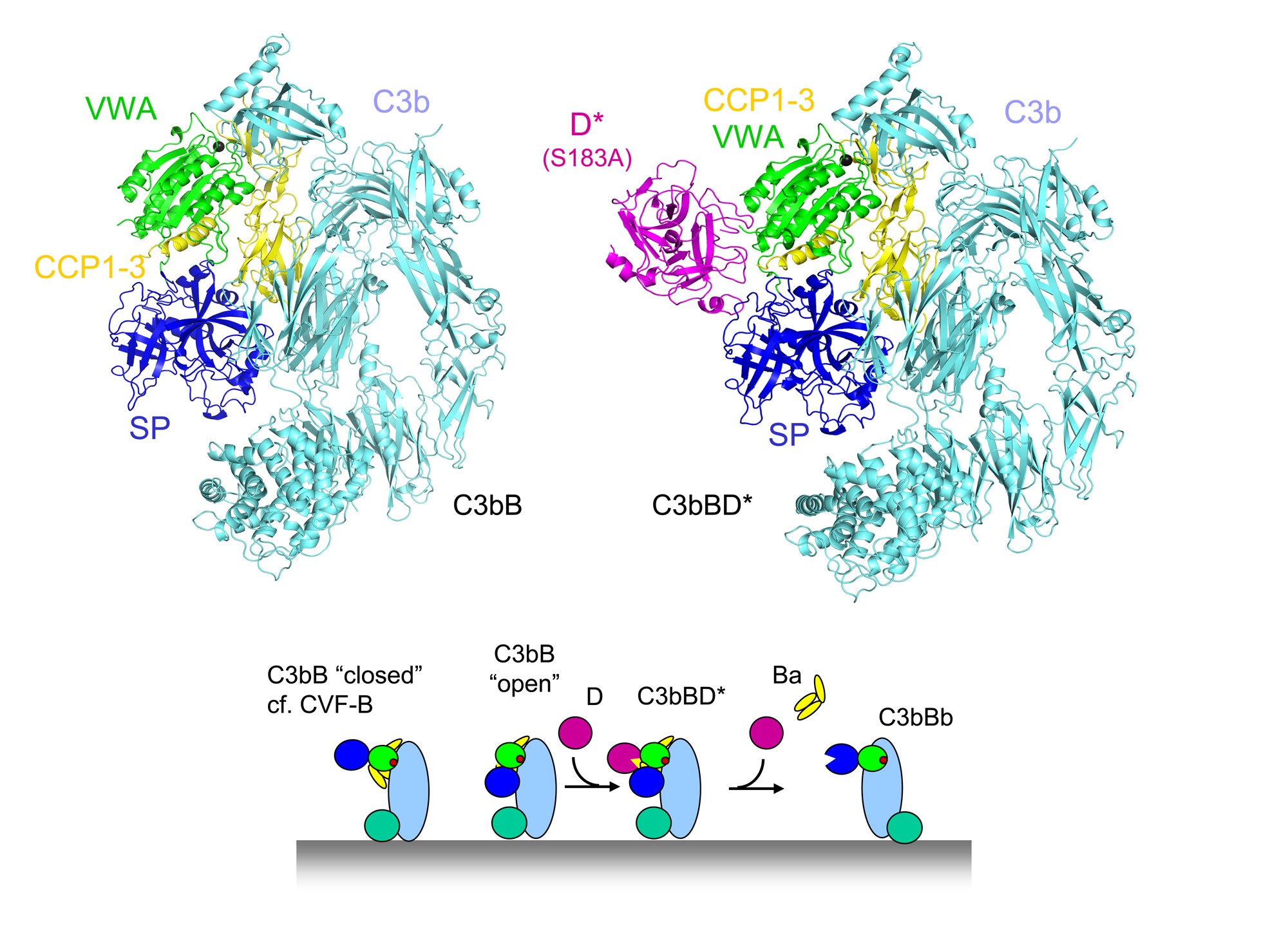
Factor B binds C3b through its Von Willebrand Factor A-type (VWA) domain and the three Complement-Control Protein (CCP1-3) domains. Binding of factor B to C3b induces a dynamic equilibrium between a closed (loading) and open (activation) form. The crystal structures of factor B in complex with the C3b homologue cobra-venom factor (CVF; Janssen et al. EMBO J 2009) and the human C3bB (Forneris et al. Science 2010) correlate excellently with the two forms observed simultaneously in negatively-stained EM grids by the Llorca group (Torreira et al. J. Immunology 2009). The conformational change from closed (loading) to open (activation) involves an unexpected, marked, reorientation of the catalytic serine protease (SP) domain of factor B. This rotation partially extends the VWA-SP linker and unwinds the C-terminal α7 helix of the VWA domain. In conjunction, helix αL that precedes the scissile bond in factor B extends by two turns positioning hydrophobic residues in pockets that are vacated by the scissile loop. Together, these changes destabilize the binding of the P1-Arginine (Arg234) to the αL and α7 helices, resulting in exposure of the whole scissile loop.
Factor D circulates in blood in an inactive conformation, in which the catalytic Ser-His-Asp triad is distorted and the P1-binding pocket is blocked by an arginine. Factor D binds factor B specifically in the open C3bB state. The tight interface formed between the "exosite" of factor D and both the VWA and SP domains of factor B explains the high affinity (Kd = 9 nM). Binding of factor D to C3bB activates factor D. Arg202, which blocked the P1-binding pocket, swings out and the self-inhibitory loop rearranges allowing the catalytic triad to be restored. What causes these conformational changes is not fully clear, though. Interaction between Glu230 of the scissile loop in factor B and Arg202 of factor D contributes to the substrate specificity and possibly provides a trigger for the conformational changes that activate factor D. Finally, factor D cleaves factor B and liberates the Ba fragment (consisting of CCP1-3 and helix αL). The proteolytic fragment Bb, formed by the VWA and SP domains, remains bound to C3b yielding the active C3bBb protease complex.
The complex C3bBb forms the active C3 convertase of the alternative pathway that (in a positive feedback loop) is responsible for the amplification of the complement response. These surface-bound protease complexes activate C3 by cleaving C3 into anaphylatoxins (C3a) that raise an inflammatory response and opsonins (C3b) that covalently mark particles for immune clearance. C3 convertases, however, are unstable and dissociate irreversibly (with a half life time of ~90 seconds for C3bBb). Thus, surface-bound short-lived C3 convertases yield a local and brief burst of complement activation.
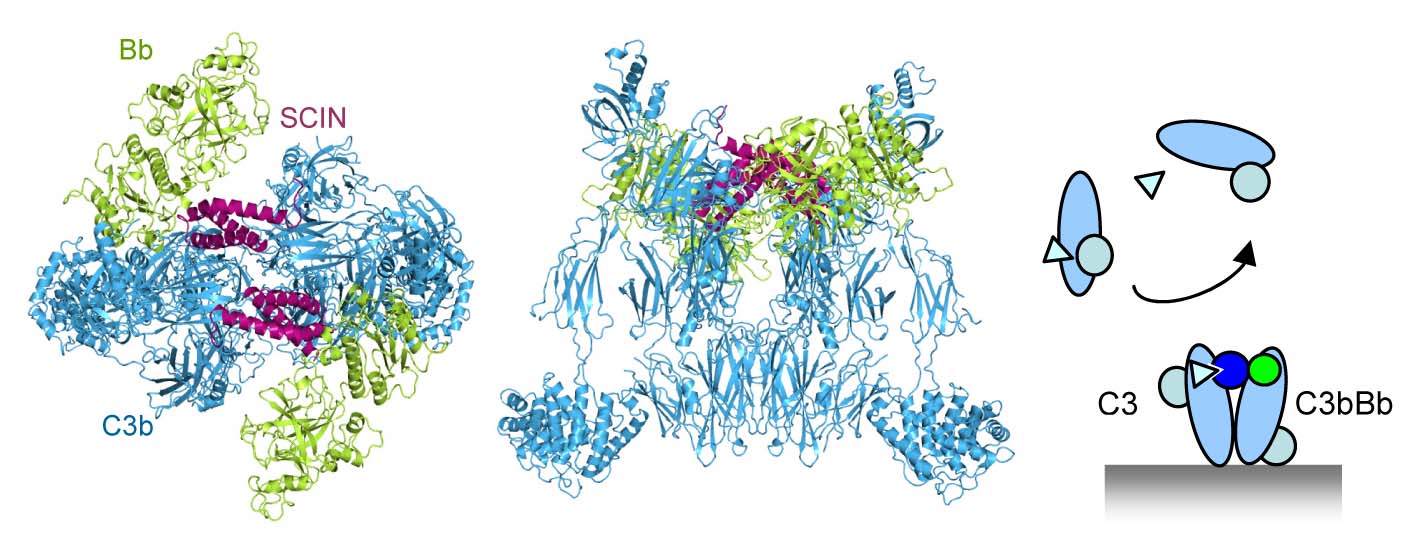
To study this labile protease complex we used an immune evasion protein, called staphylococcal complement inhibitor (SCIN), secreted by S. aureus that inhibits and stabilizes C3 convertases (Milder, Rooijakkers et al. J. Immunol. 2007). The resulting structure revealed a dimeric arrangement of convertases stabilized by two bridging SCIN molecules (Wu, Rooijakkers et al. Nature Immunology 2009). The center of the dimer is formed by C3b molecules. The protease fragment Bb is bound to the C-terminal C345C domain of C3b through its VWA domain, whereas the SP domain is oriented side-ways without making contacts to C3b. SCIN makes contacts with both C3b and Bb stabilizing the loose arrangement. The dimeric structure yielded a model for activity and specificity of the C3 convertase. The observed C3b:C3b contacts may mimic in part the substrate:enzyme complex contacts, as we predicted earlier based on inhibitor binding sites that block substrate binding (Janssen et al. JBC 2007). Replacement of C3b by native C3 positions the scissile bond of C3 in front of the catalytic site of the SP domain in Bb. A swing of the Bb fragment towards substrate would result in a productive orientation of the scissile loop in the active site; such a movement is prevented by the inhibitor SCIN. Thus, the dimerization of the substrate C3 with the ligand C3b of the C3 convertase explains the high specificity and activity of the central C3 convertases.
This model of C3 to C3b dimerization of C3 substrate binding to the C3 convertase is now confirmed by others. A dense-deposit disease mutant C3923ΔDG cannot be cleaved by the C3 convertase; however, when activated to C3b by proteases can form active convertases (Martinez-Barricarte et al. J.Clin.Invest. 2010). This is consistent with the position of the loop containing the deletion in C3 vs. C3b in the putative C3:C3bBb complex. In addition, the C3b homologue Cobra venom factor (CVF) binds its substrate C5 in the structure of the CVF:C5 complex (Laursen et al. EMBO J 2011) in an orientation consistent with C3:C3bBb dimerization model.
Properdin is an oligomeric protein that occurs in blood as flexible dimers, trimers and tetramers. Properdin enhances C3b deposition on surfaces by stabilizing surface-bound (alternative pathway) C3 convertases.
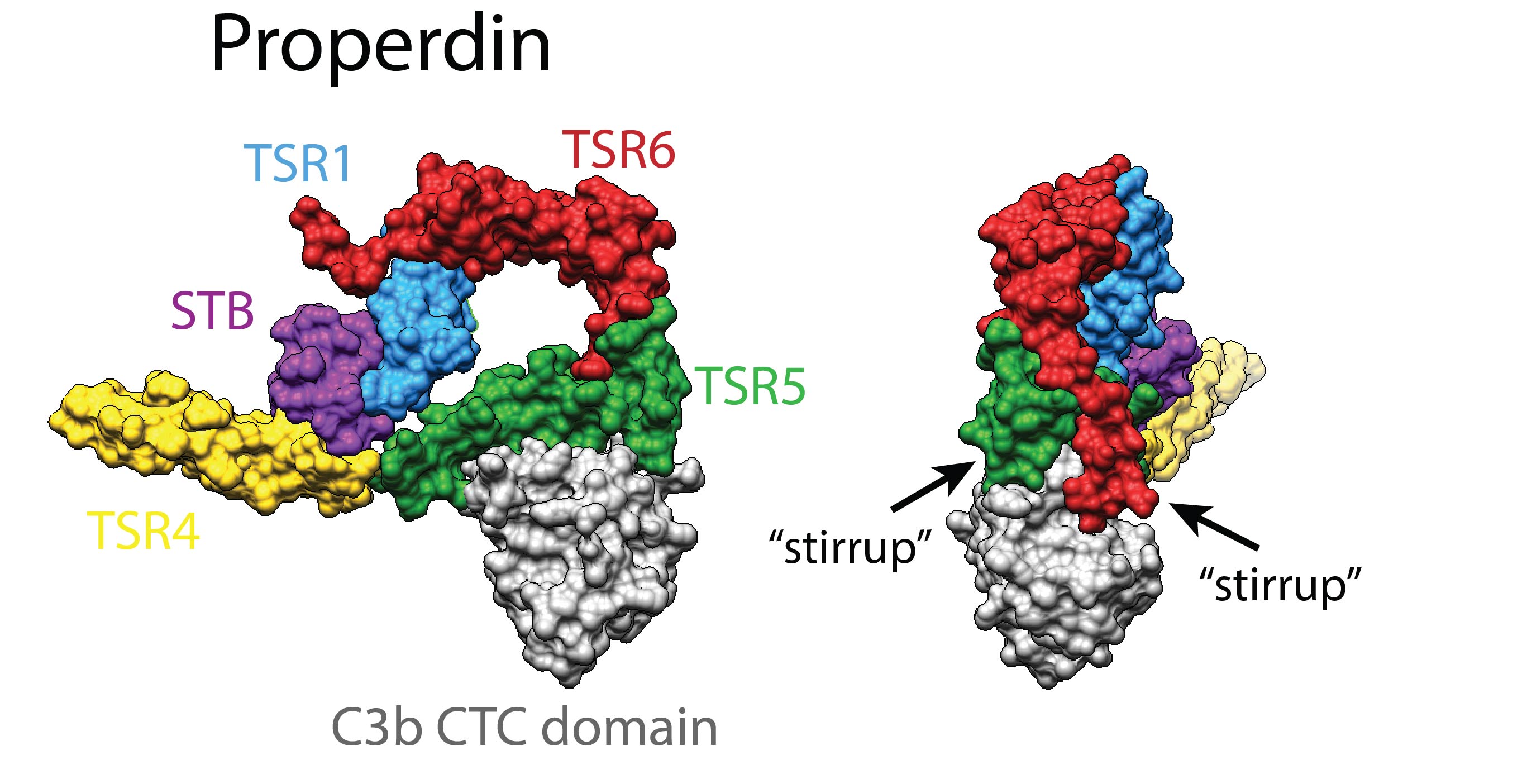
We solved the 3-dimensional structure of this heterogenic and flexible molecule by co-expressing properdin as separate N- and C-terminal constructs, thus avoiding the formation of oligomers (van den Bos et al. Front. Immunol. 2019). The crystal structures revealed a ring-shaped domain organization that was consistent with prior low-resolution data (Alcorlo et al. PNAS 2013; Pedersen et al. EMBO J. 2017). The structure of properdin in complex with the C-terminal domain of C3/C3b indicated two critical "stirrup" loops that are key to convertase stabilization. The two stirrup loops allow interactions that bridge between Bb and C3b. Flexibility in domain TSR4 and between domains TSR2 amd TSR3 allow the formation of properdin dimers, trimers and tetramers. Oligomerization directs the activity of properdin towards C3-deposited surfaces, providing 22±2 nM avidity for oligomers vs 6.8±0.1 µM affinity for single sites.
The covalent binding of C3b is indiscriminate of self and foreign surfaces. Therefore, the host requires complement regulators to protect healthy host cells and surfaces, which is a critical step as witnessed by the many mutations in regulators causing dysfunctional regulation and hence disease (typical examples of disease are: age-related macular degeneration (AMD), atypical hemolytic uremic syndrome (aHUS) and membranoproliferative glomerulonephritis type II (MPGN-II)). All regulators consist of strings of complement-control-protein (CCP) domains.
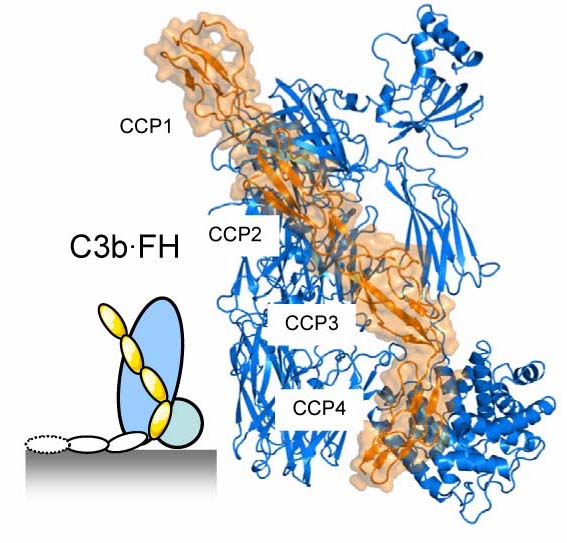
Factor H (FH) is an abundant soluble regulator that protects tissues and cells with limited surface regulators and consists of 20 CCP domains. The first 4 CCP domains are essential and sufficient for the functional activity, whereas the other domains CCP5-20 are involved in distinguishing self from foreign. We have determined the structure of FH domains 1-4 with its target C3b (Wu et al. Nature Immunology 2009). FH(1-4) binds C3b in an extended arrangement forming a 100-Å long binding site. Comparison of this structure with that of C3bBb shows that domains CCP1-2 of FH are directly involved in displacing Bb from C3b, which explains the "decay-accelerating activity" that breaks down C3 convertases and thereby stops the amplification and production of C3b. In a second mechanism, referred to as "cofactor activity", FH serves as a cofactor to bind the protease factor I (FI). FI cleaves C3b in the CUB domain yielding inactive iC3b that cannot bind factor B to form convertases and thus blocks complement amplification. Mutational data (in particular those of the homologue from variola virus done by Sahu laboratory) suggest a role for CCP2-3 in binding FI. Domains CCP2 and CCP3 lie adjacent to the CUB domains of C3b and therefore provide an appropriate binding site for FI to cleave CUB.
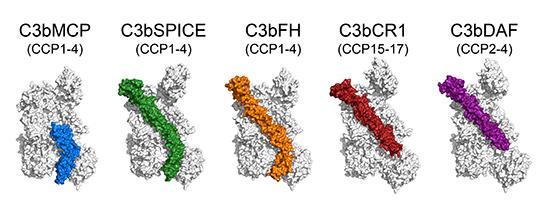
Comparison of four human and one viral complement regulators: we determined structures of human regulators MCP, DAF, CR1 and of small-pox virus SPICE in complex with human C3b (Forneris et al., EMBO Journal 2016). Together with C3b-FH (domains 1-4), these data show that the inhibitory mechanisms of decay-acceleration and cofactor activity, use one common binding site on C3b. Possibly, the two mechanisms of complement inhibition evolved from a common evolutionary origin, which has evolved into two mechanisms and a large variation of C3b-regulator interactions. Moreover, these data provide a structural framework for detailed mapping of disease-related mutations in regulators and C3b.
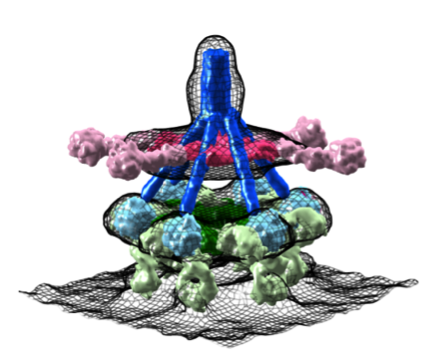
We determined the structure of Factor I (rendered inactive by mutating the catalytic serine to an alanine) in complex with C3b and regulator Factor H (using mini-FH, containing FH domains CCP1-4 linked to CCP19-20); see Figure.
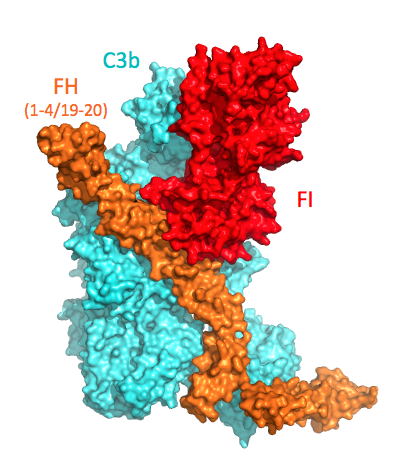
The structure of the C3b-FH-FI complex (Xue et al. 2017) revealed the critical contacts provided by FH domains CCP2 and CCP3 for binding of FI to this C3b-regulator complex. Furthermore, the C-terminal C345c (CTC) domain of C3b rotates to provide a second critical FI binding site (which is supported by a disease-related mutation Val1659Ala in C3. This site in C3b contacts the N-terminal FIMAC domain of FI, which is part of one contiguous globule formed by all 4 non-catalytic regulatory domains (FIMAC-SRCR-LDLA1-LDLA2) of FI. The extensive interactions enable FI to stabilize the conformation of its catalytic domain (cf. structure of unbound FI by Roversi et al.) and dock onto the first scissile loop in the CUB domain of C3b. Thus, the structural data and vast amount of disease-related FI mutations indicate that activation of FI is induced by stabilization of the catalytic domain, similar to what was proposed for thrombin by ligand binding to its exosites (Lechtenberg et al.).
Different regulators with cofactor activity affect the extent of proteolytic processing of C3b by FI. While, for example MCP binds C3b relatively strongly, it allows FI to cleave C3b only twice yielding iC3b. Whereas, CR1 binding to C3b enables FI to cleave C3b three times yielding C3c and C3dg. We provide an explanation for regulator-dependence of different C3b processing by FI through combining the structural insights from the ternary C3b-FH-FI complex, low-resolution SAXS and EM data of C3b and iC3b, and previously determined structures of C3b-regulator complexes (Wu et al. 2009; Forneris et al. 2016) with relative C3b and iC3b/C3c binding capacities of various regulator. As exemplified by MCP, the binding of regulator to C3b depends critically on interactions made by its CCP domains binding at positions iii and iv on C3b (using generic numbers for C3b-binding positions of CCP domains, see schematic Figure).
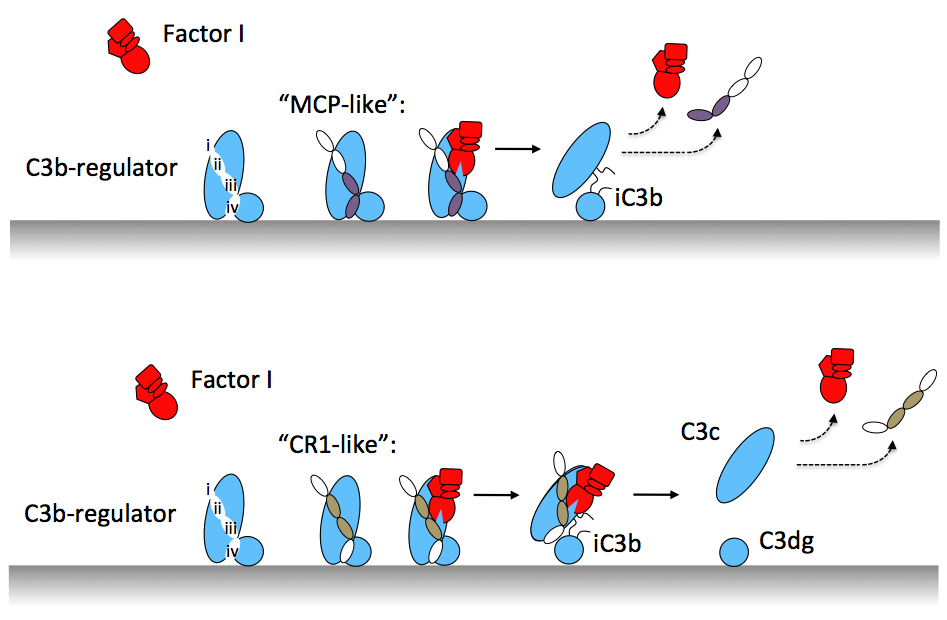
SAXS and EM data showed that only after two cleavages, the CUB domain of C3b unfolds yielding a C3c-like body of the molecule that is tethered to the membrane by the unfolded CUB domain, characteristic for surface-bound iC3b molecules. In this new arrangement, the CCPiv binding site is effectively lost, which reduces the MCP binding affinity causing disassembly of the ternary C3b-MCP-FI complex and, thereby, limiting the number of C3b cleavages to two. In contrast, binding of CR1 (through e.g. cassette III, i.e. domains CCP15-17) requires interactions made by binding at sites CCPii-iii. Upon unfolding of the C3b-CUB domain the interactions at these sites are maintained by CR1 and the regulator thus remains bound to C3b, which allows FI in the ternary complex to cleave C3b for the third time. Thus, altogether our data provide a molecular basis for understanding regulator-dependent differences in processing of C3b-opsonized cells for steering immune responses.
Activation of the terminal complement pathway results in formation of membrane-attack-complexes (MAC) that form large (100 Å wide) pores in the target membrane. These complexes are formed when C5 is cleaved into C5b by the C5 convertase. C5b binds subsequently C6, C7, (trimeric) C8αβγ and multiple copies of C9. C6 to C9 are homologous proteins build-up from a central MAC-perforin (MACPF) domain and several N- and C-terminal regulatory domains.
We determined the structure of the membrane-insertion (MACPF) domain of complement component C8α (Hadders et al. Science 2007). Surprisingly, this domain structurally resembles the bacterial cholesterol-dependent cytolysins. The resemblance strongly implies a common membrane perforation mechanism for MAC and perforin (from cytotoxic T-cells) of the mammalian immune system and these bacterial pore forming proteins.
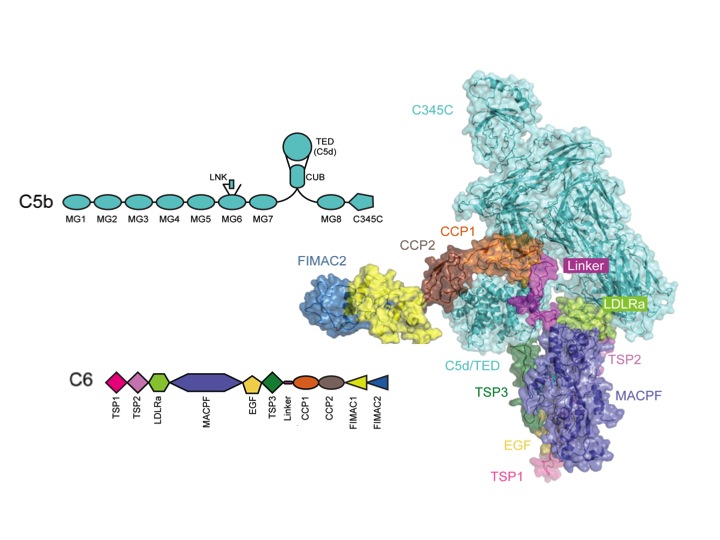
Next, we solved the structure of the C5b-C6 (C5b6) complex (Hadders and Bubeck et al. Cell Reports 2012). This structure reveals that, when C5 is cleaved into C5b, large structural changes occur. In part, these changes are similar to those of the C3 to C3b conversion. However, in the case of C5 to C5b the TED/C5d domain ends up in a position halfway the MG ring (see movie). This conformation of C5b is captured by C6. The core of C6 is bound to the bottom part of the MG ring of C5b. The C-terminal linker and CCP domains of C6 wrap around the TED of C5b. As a consequence the C6 MACPF domain with its pore-forming segments is positioned below C5b. In collaboration with the labs of Susan Lea (Oxford) and Oscar Llorca (Madrid) we placed the C5b6 crystal structure into the cryo-EM reconstruction map of the soluble MAC (sMAC of sC5b6-9). These data indicate the arrangement of C5b-C6-C7-C8β-C8αγ-C9, which forms an arc of the MAC proteins with a protrusion at the beginning formed by C5b. Below the arc large blobs of density indicate the likely presence of clusterin and vitronectin that enwrap the pore-forming segments, thereby providing protection to host cells from bystander damage.
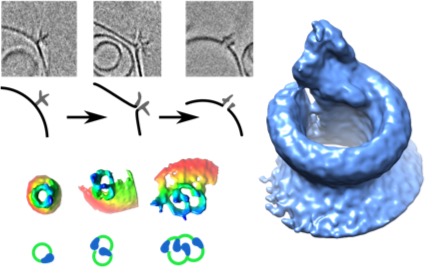
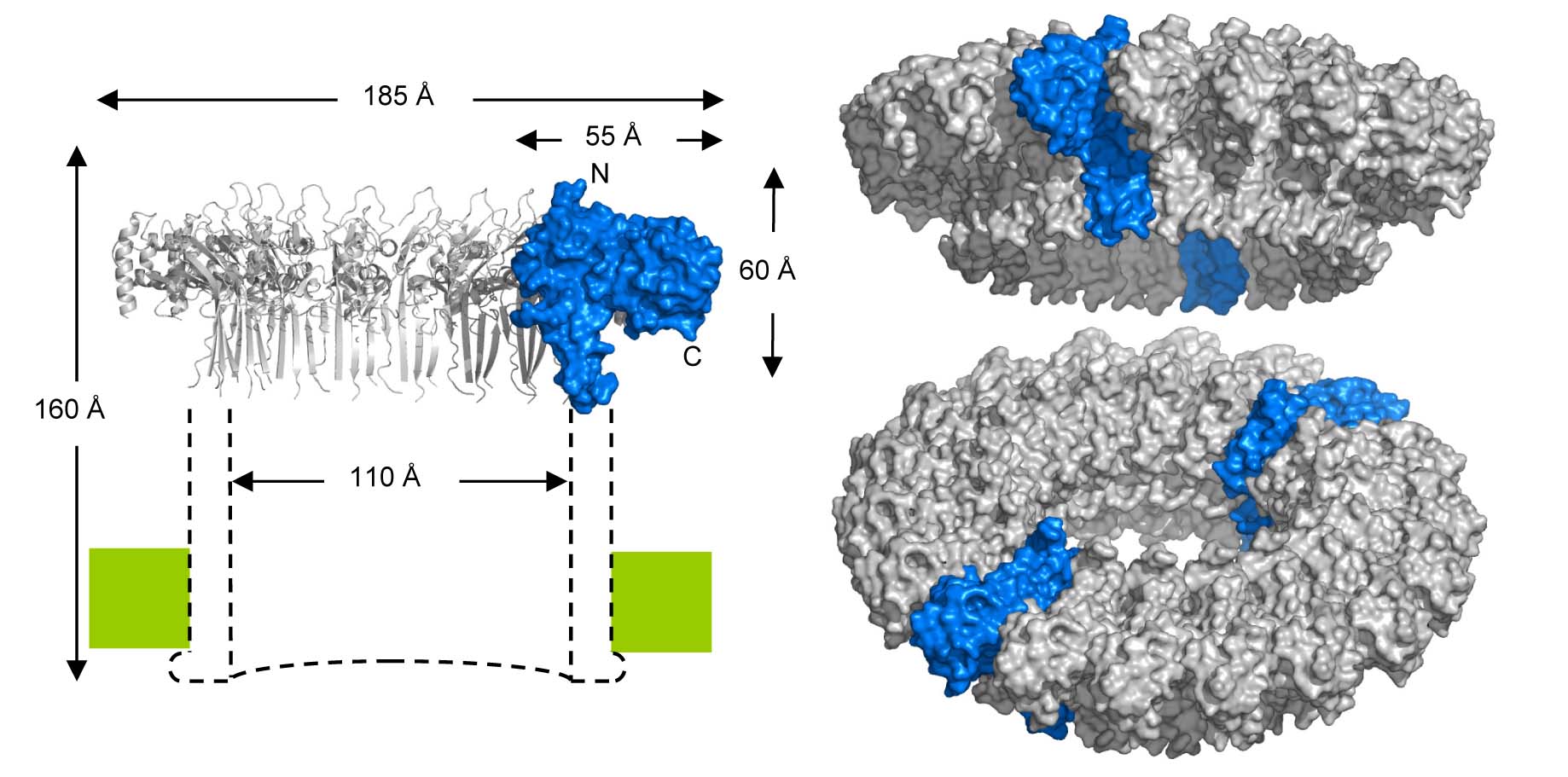
Phase-plate cryo-EM tomographic series (Sharp et al. Cell Reports 2016) revealed extensive structural heterogeneities in MAC formation. We found variable-sized oligomers for each of the C5b-7 (i.e. C5b67), C5b-8 and C5b-9 complexes on and in the membrane. Complexes of C5b-7 adhere to bilayers, bulging out the outer-leaflet, while maintaining the integrity of the membrane inner-leaflet (see Figure). In contrast, oligomeric structures of C5b-8 perforate the bilayer, forming a oligomeric C5b-8 pore, explaining that membrane leakage is observed in the absence of the ring-forming C9. C5b-9 (MAC) rings were observed forming both single pores and multimeric pores of various shapes. Reconstruction by sub-tomogram averaging yielded a 20-Å resolution structure revealing a twisted, slightly cone-shaped MAC pore in the lipid bilayer. Poor closure between the C5b-8 initiator and the C9n-end yields a remarkable seam in the barrel above the membrane. Most likely, the twisted, cone-shaped barrel with its seam is due to C9 compensating for the missing, or shortened, transmembrane β-strands of C6, C7 and partly C8. Moreover, the irregular closure explains the observed variation of ring closures associated with multimeric C5b-9 pores. These data indicate a dynamic process that leads to formation of MAC pores. Possibly, the dynamical and variable process allows the MAC proteins to attack a variety of membranes presented by diverse pathogenic microbes.