THE UPACK PROGRAM PACKAGE
Bouke P.
van Eijck, Department of Crystal and Structural Chemistry, Bijvoet
Center for Biomolecular Research, Utrecht University, Padualaan
8, 3584 CH Utrecht, The Netherlands. E-mail: vaneyck@chem.uu.nl
or vaneijck@xs4all.nl.
Version
1, 1994. Specially written for hexapyranoses
Version 2, 1996. General molecules, more space groups possible,
polarization.
Version
3, 1998. Code-directed input, automatic threshold setting, force field through
code numbers.
Version
4, 1999. More than one independent molecule possible.
Version
5, 2000. Treatment of lattice vibrations possible.
Version
6, 2004. Automatic reduction of high-symmetry space groups to lower symmetry.
Version
7, 2006. Charges on non-atomic sites possible.
Version
8, 2010. Complete update.
Version 9, 2011. Topology structure improved.
Version 10, 2012. Rigid structures possible, double precision throughout.
Version 11,
2016. Improved energy window settings for large molecules.
Version 12,
2020. Possibility to exclude knotted structures.
Version 13.2,
2021. Bugs removed (and possibly new ones introduced).
Contents
Output files
Simple force field
Detailed instructions for the preliminary stage
Detailed instructions for the search stage
Update November 2015.
The sixth blind
test of crystal structure prediction (October 2015) has led to new insights.
See Ref. 1b and the supplementary material referring to
UPACK.
Concerning
UPACK, the structure generation became intolerably slow for the large molecules
considered in this test. It was found that here the windows determining
acceptance or rejection of possible structures, as optimized for small
molecules, were too small in several stages of the procedure. It concerns the
settings of ebig and emax1
in pack12 and emax1 in pack.3 In this
version their values are set as function of the number of atoms. These settings
can be overruled manually; it may be useful to experiment and report possible
improvements. This is especially important for structures with more than one
independent molecule.
Concerning
structure prediction generally, the problem is judged by some participants to
be essentially solved. For details see the publication on this blind test.
Briefly, both structure generation and ranking should depend on DFT-D
calculations. Large clusters of processor units are needed, using over 100000
computing hours per structure.
Thus UPACK
is essentially reduced to being a cheap way of getting some idea of the energy
landscape, sometimes hitting on the correct minimum and sometimes being way
off. For more reliable results the best UPACK structures should be used as
starting point for better calculations. An affordable extension is to use
individual ab-initio point charges and intramolecular energies.
Some UPACK
features, attractive as they seemed at the time, have proven to be hardly
helpful:
- Polarization
- Molecular dynamics
- Charges on non-atomic sites
- Vibrational corrections
- Grid search
Their use
is not recommended.
Introduction
The program
package UPACK (Utrecht Crystal Packer) can construct hypothetical crystal
structures of low potential energy, using a molecular force field. It was
originally designed to study a family of compounds, e.g. carbohydrates, with
one and the same force field. For details of some force fields implemented in
UPACK see the force field manual. Meanwhile it
has become clear that such an approach is hardly useful because it is necessary
to develop an individual force field for each molecule studied. What UPACK can
do is only the creation of a list of structures which probably contains those
that are experimentally feasible. For a reliable ranking more sophisticated
programs are needed. However, the methods to go beyond empirical force fields
are not implemented in the standard program package. For extensions see the ab initio manual. Other investigators have developed
different approaches, of which the DFT-D methods appears to be the most
promising one. Before deciding to use UPACK, the reader is urged to study the
recent reports on blind tests on crystal structure prediction [1]
and references therein for background information. See also some lecture notes,
which can be viewed immediately in PDF format
or downloaded.
In the older versions of UPACK a systematic grid search was used [2].
However, we have found that a random search performs at least equally well, and
is easier to handle in practice [3]. Therefore, the main
part of this manual covers only the random search method, and the grid search is treated separately. Both approaches are
fairly time-consuming, and require a simple force field with a low cutoff
radius for nonbonded interactions. At the end a preliminary list of possible
structures is obtained.
The program suite UPACK
(Utrecht Crystal Packer) is made available under the following conditions:
The programs are available free of charge for
academic use only, excluding commercial purposes.
In publications where UPACK has been used
acknowledgment must be made to the author.
No responsibility is taken for errors in the
codes or documentation.
Structure
generation is restricted to space groups with triclinic, monoclinic or
orthorhombic symmetry. The programs are under constant development. Please
report any problems encountered, including the version date.
The substance to be studied
is characterized by a group of separate molecules. Such a group is the smallest
possible repeating unit, for instance ABB in a dihydrate where A is a certain
molecule and B is water. The constituent molecules are building blocks for the
generation of hypothetical crystal structures. Generally several distinct
molecular conformations are thinkable. It is possible to specify their
geometries precisely, and to investigate a list of those that are deemed likely
to occur. Alternatively, one may rely on automatic generation of possible
conformations. They are defined by the dihedral angles (symbol ω)
around single bonds. We shall denote the number of such bonds by Nz and the number of less essential
conformational degrees of freedom (for instance methyl groups) by Nh.
In the present version of UPACK this distinction is not essential, it is
retained for compatibility.
Furthermore, let the group
contain Mg molecules and Ag atoms. Of
course, all these properties of the group are independent of the actual
molecular conformation and crystallographic environment.
To generate a crystal, a
number (G) of independent groups is placed in a certain space group
which has S general equivalent positions. We shall exclude the
possibility that a symmetry element of the group coincides with a symmetry
element of the lattice: such a structure must be described in a space group
with lower symmetry, disregarding that symmetry element. Thus the G groups
always coincide with the asymmetric unit, and the total number of groups is S
× G. The number of really nonequivalent molecules in a unit cell will be
denoted by Z''. In the example of the dihydrate ABB it can be seen that
Z'' must be between 2 and 3G.
Possible crystal structures
are generated by placing molecules with varying positions and orientations into
a unit cell of varying dimensions. This gives at most twelve (P-1) and
at least eight (e.g. Pna21) parameters to be varied; this
number can be considerably larger if the molecules are flexible or if more than
one independent molecule must be placed. One of the cell parameters can be
eliminated by using an approximate crystal density.
The calculations must be
repeated for all probable space groups. Fortunately, relatively few space
groups account for about 90% of the actually observed structures for organic
molecules with Z'' = 1: P21/c, P-1, P212121,
P21,Pbca, C2/c, Pna21,
Cc, Pca21, C2, P1, Pbcn,
P2/c. If sufficient computer power is available, one may extend the
coverage by including P21/c, P1, P-1, P212121,
P21 with G = 2 [4]. If the
substance is chiral, only P1, P21, P212121,
and C2 remain. For a group with internal symmetry the generated
structures often belong to a space group with higher symmetry. The
determination of the actual space group of these structures is not possible in
UPACK; programs like PLATON
[5] may be employed.
Except for input and
output, where fractional coordinates may be used, the programs work with
Cartesian coordinates in nanometer units. The crystallographic a-axis
coincides with the Cartesian x-axis and the b-axis is positioned
in the x-y plane. The crystallographic axes are projected on the
Cartesian axes and the components ax, by, cz, bx, cx
and cy are used to characterize the unit cell.
The crystal is built from
rigid molecules, defined in a local axes system with aid of three reference atoms
for each molecule. The axes system is chosen in such a way that:
the plane through these three atoms is parallel
to the x, y-plane,
the vector from atom 2 to atom 1 is parallel to
the positive x-axis,
the y-coordinate of atom 3 is larger
than the y-coordinate of atom 2,
the origin coincides with the average
coordinates.
The
position of this molecule in the crystal is determined by translation and rotation
of the local coordinate system. More precisely, any atomic position vector (x,
y, z) in the crystal is found by rotating the corresponding
vector (x0, y0, z0) in
the free molecule over an angle φ around the z-axis, over
θ around the x-axis, over ψ around the z-axis, and
finally translating over X, Y, Z:
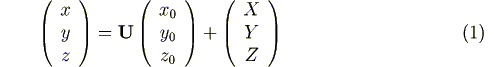
with
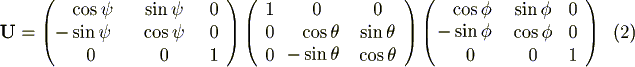
Energy calculations
Energy minimization is the
central technique of the method. The energy is always reported in kJ/mol for
one group (although other units may be used in the force field files). The
programs were developed from routines from the GROMOS molecular dynamics
simulation program package [6] as basis, and we are grateful
to Professor W. F. van Gunsteren for his permission
to do so.
As discussed above, in the
searching stage fast calculations are essential. A Lennard-Jones 12-6-1
potential is used here. Ewald summation may be used for the Coulomb and attractive
r -6 dispersion terms, and for further work a Buckingham-type
exponential repulsion is often preferable to the r -12 term.
Atomic polarizabilities may also be included, although we have not found that
refinement very helpful.
To allow fairly reliable
calculations without an extremely large cutoff radius, the molecules can be
split up into charge groups. The cutoff criterion is based on charge groups
rather than on individual atoms. If the charge groups are chosen to have
(almost) zero net charge, the electrostatic interaction decays at long distance
with r -3 rather than r -1. To allow for
small errors in the force-field setup, the total molecular charge is set to
zero by adding equal amounts to each atomic charge. Alternatively, convergence
acceleration can be used for Coulomb interaction and r -6 van der Waals attraction.
The energy minimizations
are performed by a number of steepest descents steps, followed by a more or less
exhaustive conjugate gradients procedure. The use of pair lists for nonbonded
interactions (as well as lists of vectors in reciprocal space for Ewald
summations) is essential for stable convergence [7]. These
lists are only updated upon a significant change of any geometric parameter.
Unavoidably, the same
structures will be encountered repeatedly, probably with different choices for
the unit cell parameters. Of course, it is very important to eliminate such
equivalent structures by a clustering procedure. A general algorithm, based on
comparison of radial distribution curves, is available.
Installation
You can
obtain the program via anonymous ftp. It is distributed in the form of a
compressed tar file (download).
In this manual file names are given in brown color.
Instructions to be given for executing the script files are red.
The following directions
apply to Linux systems. Little testing on other operating systems has been
done, but all programs are in simple Fortran77 and should be fairly portable.
Deviations from the standard are:
the use of include files.
the use of the do ... enddo
syntax rather than labels
the specification of some integers: two bytes
for memory reduction, four bytes if necessary.
the use of ANSI-type color codes (in case of
problems replace color.i:
cp no-color.i
color.i).
In previous
versions single precision was preferred for the first stage of the structure
generation to obtain greater speed, but the difference seems to have become
negligible on modern computer systems. Now double precision has been
implemented throughout; this is absolutely needed in the final stage to obtain
full energy convergence, which also increases the reliability of the clustering
of equivalent structures.
The program package is
normally delivered in a tar archive: upack.tar.gz.
Decompress the tar archive:
gunzip upack.tar.gz
Unpack the resulting file in your home directory:
tar xvf upack.tar
This should create a directory upack
directly under your home directory, with subdirectories progs, scripts, tops, data, upackdoc and abinitio. Existing files
with the same names will be overwritten, others will remain untouched.
The subdirectory progs contains:
the programs and subprograms (all starting with
the letter y)
the include file params.i, which contains most array
dimensions (to be changed if required by
your needs)
the other include files (all starting with the letter c
and having the extension .i)
containing the common blocks
various script files for compiling and linking, assuming the Fortran compiler gfortran
In case gfortran is
not available, copy all files from subdirectory f2c-gcc
to replace the corresponding ones in progs and
try again; now f2c and gcc are used. This may help,
but the corresponding executables are probably slower.
The
subdirectory scripts contains various script
files to run the programs (all starting with run)
and some other utilities. The scripts runpa12, runpa1, runpa2, runpa3, runpp and runmd end with printing "Computer name",
which you may want to replace by your own computer name for later reference.
To be able to call the scripts from other directories, it is very convenient to
have upack/scripts in the path.
To compile and link the
programs, go to subdirectory progs and call
compall
This should create the executables (*.e). An individual
program or subroutine may be recompiled after a modification, e.g.:
f yprep
linkprep
The
subdirectory tops contains
the force field files (ending with .ff), the codes files (ending with .cod), and the space group
file all.spg.
The subdirectory upackdoc contains the actual version of this manual.
The subdirectory abinitio contains a framework to combine UPACK with ab-initio
calculations. Not all necessary programs are included for copyright reasons;
see the ab initio manual.
The
subdirectory data
is meant to contain your data files. It should contain a subdirectory subst for every substance to
be investigated, and here the topology file(s), e.g. subst.tps, should be placed. The
best policy is to create sub-subdirectories for every space group to be
considered. If you wish to use experimental data for comparison, they should be
placed in a sub-subdirectory exp.
Some
subdirectories under data are
already included for demonstration and testing purposes. The example for the
random search is ethanol which has two independent
molecules in space group Pc. It is an exceptionally lucky case where the
experimental structure is found quickly. Another simple example is benzene where a complete structure generation is
demonstrated. The last examples are in data/glucsa, which is meant to
demonstrate the now hardly ever used grid search,
and in data/methyd for the ab initio option.
List of files containing programs
|
ycodes.f |
Creates framework
for topology files |
|
ypack1.f |
Generates
structures with one independent molecule in grid search |
|
ypack2.f |
Generates
hydroxyl torsional angles in grid search |
|
ypack12.f |
Generates
structures with more than one independent molecule |
|
ypack3.f |
Minimizes the energy |
|
yclus.f |
Removes
equivalent structures with one independent molecule |
|
yzoek.f |
Searches
for one equivalent structure with one independent molecule |
|
ydist.f |
Combines
the latter two functions for the general case |
|
yprep.f |
Minimizes
the energy for one structure |
|
ycrysmd.f |
Performs
molecular dynamics calculations for one structure. |
|
ymktop.f |
Creates
topology file with force field values. |
List of files containing subroutines or functions
|
ybonde.f |
cbond, choek, cdiha: calculates intramolecular energies |
|
ycart.f |
cart:
constructs coordinates for a free molecule |
|
ych.f |
ch:
calculates eigenvalues and eigenvectors (from EISPACK) |
|
ycongra.f |
congra:
performs conjugate gradients energy minimization |
|
yconn.f ycatena.f |
conn:
finds connected atoms from coordinates for a free molecule catena:
enables the treatment of knotted structures |
|
ydclus.f |
dclus:
compares possibly equivalent structures |
|
ydens.f |
dens:
estimates initial density for pack12 |
|
ydiff.f |
diff:
compares possibly equivalent structures |
|
ydihan.f |
dihan
(function): returns the value of a selected dihedral angle |
|
ydlist.f |
dlist:
makes list of nonbonded distances for dclus |
|
yexpsym.f |
expsym:
expands symmetry operations to the full group |
|
yfiles.f |
files, fdat: makes mdi, spf, xyz, cif or dat
type of coordinate file |
|
yfunc0.f |
func0:
calls energy routines for a free rigid molecule |
|
yfunc1.f |
func1: calls
energy routines for a rigid molecule in the crystal |
|
yfunc2.f |
func2:
calls energy routines for a rigid molecule with rotatable hydroxyl groups |
|
yfunc3.f |
func3: calls
energy routines for a flexible molecule in the crystal |
|
ygeom.f |
geom:
calculates and compares geometric parameters |
|
ygetsym.f |
getsym:
finds all relevant symmetry elements |
|
yident.f |
ident:
rearranges and relabels coordinates to conform with molecular topology |
|
yintre.f |
intre: calculates intramolecular energy |
|
yintrot.f |
introt:
sets up data for internal rotation |
|
ykwald.f |
kwald:
calculates reciprocal part of Ewald correction |
|
ylatvib.f |
latvib: calculates lattice vibrations |
|
yleesc.f |
inpt (function):
reads and stores card-directed input |
|
yleesmd.f |
leesmd:
reads data for molecular dynamics run |
|
yleesr.f |
leesr:
reads general space group information |
|
yleess.f |
leess:
reads coordinates and expands equivalent positions |
|
yleest.f |
leest:
reads molecular topology information |
|
yleesx.f |
leesx:
reads geometry information, places hydrogen atoms |
|
ylist.f |
list:
calculates nonbonded energy and forces from pair list |
|
ylistk.f |
listk:
calculates reciprocal part of Ewald correction from list |
|
ylistp.f |
listp:
as list, with polarization |
|
yltw.f ynewchg |
nword
(function), realstr, intstr:
internal read procedure newchg:
reads and expands individual charges |
|
ynonbe.f |
nonbe:
calculates nonbonded energy and forces |
|
ynonbep.f |
nonbep:
as nonbe, with polarization |
|
yopth.f |
opth, listh: calculates energy for various hydroxyl torsional
angles (in grid search) |
|
yrunmd.f |
runmd, stopcm: performs molecular dynamics run |
|
ysecder.f |
secder:
calculates second derivatives for lattice vibrations |
|
yshake.f |
shake:
keeps bond lengths constant during MD run |
|
ysort.f |
sort:
sorts data in ascending order |
|
yspepos.f |
spepos:
tests for special positions and reduces the symmetry if necessary |
|
ystedes.f |
stedes:
performs steepest descents energy minimization |
|
ysubs.f |
random
(function), norm, prod: various utilities |
|
ytest.f |
test:
numerical check on calculation of derivatives |
|
ytrans.f |
trans:
transforms to standard axes system |
|
ytransf.f |
transf,
matmat, invbox:
transforms to another crystal setting |
|
ytri.f |
tri:
transforms to oblique coordinates and back |
Overview
The general
course of a crystal structure prediction is as follows.
1. For every substance the
molecule(s) in the group must be defined through a topology file.
Originally this file contained code numbers for every force field contribution
(like bond angles, atomic charges, and so on). This scheme was designed to
treat a large number of related compounds with the same force field. Nowadays
it is obvious that such an approach is bound to fail, and that a force field
should be designed for each compound individually. For that purpose it is
easier to include bonded force field terms and charges directly in the topology
file, and this is now the standard.
2. For every conformation
to be studied separately, a coordinate file for the constituent free
molecule(s) has to be prepared. Note that often one input conformation can
suffice, since rotational freedom about single bonds can be taken care of automatically.
3. The structures are
generated by a random search for one or more independent molecules by the
program pack12. Each molecule is placed with random
orientation, random position, and (possibly) random rotatable dihedral angles
in a crystal with random cell axes and cell angles. The energy of the generated
structures is provisionally minimized with either rigid molecules or full
geometry relaxation. A Lennard-Jones type force field
is recommended here to avoid infinitely negative energies which can occur in
exponential repulsion when atoms are placed close together.
4. The results are
clustered by the program dist. This program
also determines the number of crystallographically
independent molecules (Z'').
5. The minimization is
continued with the program pack3. A first
cycle could be done with different settings for the same force field as used in
the search, or another force field may be preferred. After each cycle the
results are clustered again.
6. Collecting and
clustering the results from the different space groups delivers a list which
may be considered as final, but preferably used for further study in a
subsequent stage with a more sophisticated force field. There is the
possibility of including polarization, relaxing the space group symmetry by
expanding to P1, and to perform a primitive molecular dynamics
"shake-up". In this way the stability of the obtained crystal
structures can be investigated. It is also possible to study the effect of
lattice vibrations on thermodynamical quantities [8].
Preferably, using other program packages, atomic multipoles and/or ab-initio
energies should be used. A word of warning: it cannot be taken for granted that
there is a one-to-one correspondence between the crystal structures in
different force fields.
If the experimental
structure is known, one will want to know whether or not it was found by the
packing procedure. The program dist can be used for this purpose too, if the
experimental structure is first energy-minimized with the program prep.
Molecular dynamics
calculations can be carried out individually for one structure with aid of the
program crysmd.
Knotted structures (addition 2020)
The possibility of detecting
catenated rings ("knots") has been introduced. These may be generated
in structures with large rings; they are unrealistic unless specifically
intended and defined as such.
The codeword knot allows this check in three programs:
pack12 and pack3:
all such structures are eliminated from the search results.
prep: a suspected structure can be
analyzed in detail.
Test calculations suggest that it is
probably better not to set knot in pack12, but to remove them later by using pack3 or prep.
Somehow these two approaches do not always give compatible results; indeed, the
algorithm developed has some dubious features. Suggestions for an alternative
will be welcome.
This feature needs a file rings.def to define the rings. If absent, the program
tries to produce it. Please check the results. In any case, this does not work
(yet) in the presence of a C atom which is part of 4 rings.
Files for input and output
have fixed names in the various programs, to be described in detail below.
These file names should be linked to actual files present on disk. To this end
a number of C-shell scripts is available which link the necessary files and
execute the programs. In this manual a certain convention for directory structure
and file names will be used; other users may prefer to create a different
system.
File names are given in brown color. Instructions to be given for executing
the script files are red. Many programs require code-directed
input: on each input line a code word is
followed by a number of numerical values which are identified by colored names. To aid in understanding the Fortran
sources, these names generally (but not quite always) correspond to identifiers
used in the programs. The symbol # indicates that a certain number must be
inserted in that place.
A script file to execute a
certain program typically looks like this:
runprog s/a/... [filenum] subst [coord] ffname
Here runprog is one of
the execute scripts, to be discussed in detail later. Some or all of the
following parameters must be specified; entries between square brackets [...]
are optional. A short explanation:
s/a/... : select the appropriate topology file,
defined by the extension in subst.tps,
subst.tpa, ...
filenum: a number (#)
referring to a pack-type file (vide infra).
subst: a chosen name
for the substance under study, which is also the directory name.
coord: the name of a
coordinate file.
ffname: the name of
the force field file ffname.ff to be used.
Most input files must have
a precisely defined structure, but the ones that specify the course of the
calculations are code-directed: parameters have default values unless specified
by a line ("card") starting with one of a well-defined set of code
words. An overview is given here, while a detailed description is given in a
later section. Some explanation is also given as comments at the beginning of
the subroutines.
The following files are
more or less general, and have to be updated only incidentally:
all.spg: data on space group symmetry.
codname.cod: defines atom types and code
numbers for various terms that contribute to the potential energy.
ffname.ff, various force field files to
provide the actual numerical parameters corresponding to the code numbers.
For every
substance the following files must be prepared:
subst.tps, subst.tpa: the topology file. The program codes may be helpful here; it requires the preparation
of a file subst.con which gives the molecular connectivity as well as
the atomic charges. The topology file may now also contain the bonded force
field parameters and the charges.
cor.# (# = 001, 002, ...): atomic coordinates for
every molecular conformation of the group to be packed. These files may be
prepared by any relevant program; in UPACK the program prep
can be used which will ask for the values of all dihedral angles.
Optionally,
the default specifications can be changed by entries in the files:
subst.pa# (# = 1,2,3,...): input specifications for the
various stages in the crystal structure generation. These files may be omitted
if the default specifications are adequate.
For
calculations on single structures we also
need:
coord: crystal cell data and atomic
coordinates.
subst.pp: input specifications for prep.
Note: input files prepared in
Microsoft editors are often misinterpreted by Linux systems! Re-editing in
EMACS may help.
The generated structures
are collected in parameter files pack.#, the
number # indicating the stage of the calculation in which the file was
prepared:
# = 10: first stage (from pack12)
# = 20, 30, ...: subsequent stages (from pack3)
These files contain general
information about the generated structures. Detailed atomic coordinates are
delivered in corresponding files pack.19, pack.29, ..., respectively. These files generally
contain equivalent solutions, which can be removed by the clustering program dist. This increases the file numbers by 3 (e.g., pack.30 is clustered to pack.33).
It is also possible to
produce individual coordinate files of the hypothetical crystal structures in
formats mdi, spf, xyz, cssr, cif or fdat (for further use in UPACK or other
packages).
General output information
is given on a file out.pri,
which serves as a record of the simulation conditions. This file is usually
linked to pack.pr#, distcl#,
and so on. There is also output to the screen to signalize error conditions,
and to obtain (optionally) more output for debugging purposes.
Since general
force fields are of limited use, we may just as well use a very simple force
field to construct a first version of the topology file, to be improved
manually for each individual compound. It takes care of the most frequently
occurring atom types:
|
HA |
H in alcoholic OH |
|
HZ |
H in carboxylic OH |
|
HN |
H on nitrogen |
|
HC |
H on aliphatic C |
|
HR |
H on aromatic C |
|
HW |
H in water |
|
HD |
H on
double bonded C |
|
CP |
C with single
bonds (tetraedral) |
|
CD |
C with
one double bond |
|
CR |
C in aromatic ring |
|
NP |
N with
three bonds (pyramidal) |
|
ND |
N with a
double bond |
|
NR |
N in aromatic ring |
|
OD |
O with double bond |
|
OA |
O in alcohol |
|
OZ |
O in carboxylic OH |
|
OW |
O in water |
|
OS |
O in ether |
|
SD |
S with double bond |
|
SR |
S in aromatic ring |
|
F |
|
|
Cl |
|
|
Br |
|
|
I |
|
Nonbonded parameters in the force field are from the OPLS Lennard-Jones
potential (simple-lj.ff as in opls.ff) or from the Price-Williams Buckingham
potential (simple-pw.ff as in will.ff); energy units are kcal/mol, distance units
Angstrom. Torsional parameters are only set for aromatic systems; other ones have to be determined
for each compound individually. Modern force fields
depend on atomic charges from ab-initio quantumchemical
calculations. For demonstration purposes only, the simple force field contains
charge increments for end atoms (H, O) with some general usefulness. Of course,
for real applications this is inadequate. Moreover, aromatic and conjugated
systems are unique in principle: geometry parameters and charges have to be
derived individually from theoretical chemistry.
Only nonbonded
force field terms are needed when the topology file
contains also the information for bonded contribution and charges. Then
a "stripped" force field file is sufficient and can be used nearly
universally. To this end there exist the files lj.ff and pw.ff.
Procedure
For
each substance all work should be done in a subdirectory data/subst. The detailed formats
for the various input files are given in later sections.
1. Prepare the connectivity file(s) subst.con. This is helpful to
obtain the molecular topology file(s) by running the program codes:
runcod subst.con [codname]
using a codes file codname.cod. The
default codname is simple. Alternatives are opls and, for united atoms, unitat. If an atom
type is missing, update the codes file and try again.
2. The output
file out.tp
can, after editing, be used as topology file.
However, as discussed above, it is easier to have a topology file that
includes bonded force field terms and charges for a specific molecule. For that
purpose an auxiliary program mktop is now available:
runmk out.tp [forcefield]
using a force field file forcefield.ff. The
default forcefield is simple-lj. This program
produces the output file out.up
which can be renamed to subst.tps (but see the next paragraph).
An additional new feature is that mktop
can also use external charge input, e.g. from an ab-initio calculation. This
information can be given in either .cssr format or .xyz format, and the relevant file should be copied to
charges.in. All previous charge information
from force field or topology is discarded.
3. The output
file out.up is
a framework which usually needs editing. It is immediately necessary to inspect
out.up for the warnings ***** which indicate the absence of a vital
force field parameter. Replace the zero values by appropriate ones and check
for further possible improvements. The simple force
field produced in this way is good enough for testing purposes, but obviously
inadequate for real work. So extensive further editing is necessary, especially
for charges. Rename the edited file to subst.tp# (e.g. subst.tps for the simple force
field, but replaced by subst.tpa, subst.tpb, ... in later improvements).
4. If
necessary, update the space group file all.spg.
5. Decide which
conformations are to be used for crystal packing, and prepare a coordinate file
cor.# for each of them (# =
001, 002, 003, ...) for a free molecule. To create a coordinate file many programs
exist. In UPACK it can be done by running the program prep (for details see below).
However, more often than not this option fails. It is better to use a graphical
program, for instance MOLDEN.
This program can produce an .xyz-type coordinate file
which can be directly read by UPACK.
Decide which
space groups are to be investigated. Avoid Buckingham-type force fields in this
stage, as infinitely negative energies can occur when atoms are placed close
together. Create subdirectories named after these space groups (with omission
of slashes, e.g. subst/p21c). In every subdirectory a search must be carried out.
Usually the default settings are adequate; otherwise input files subst.pa# must be created. Copy
the coordinate files cor.#
into each subdirectory. The random search consists of the following steps:
1. Generating
and clustering structures:
runpa12 a subst forcefield
Output files are pack.10
(parameters) and pack.19 (coordinates);
the parameter file must be clustered to pack.13:
rundist a 10 subst
To save computer time a united-atom force field and/or a rigid-molecule
search might be used. This is generally not recommended, and should only be
done as a last resort.
2. Energy
minimization: for instance, to continue with another force field:
runpa3 a 13 subst opls
rundist a 20 subst
with final results on files pack.23 and pack.29.
The program prep can be used to minimize
the energy of a given individual crystal packing. The crystal cell data and
atomic coordinates are given in an input file coord. The space group information
can be taken from this file or communicated through a codes file subst.pp. Run the program:
runpp a subst coord
forcefield
Output coordinate files are given in various formats.
An important
application is the minimization of the energy of an experimental polymorph, if
available. The geometry changes give an indication about the quality of the
force field, as given by the parameter D. Also, the energy-minimized
experimental structure should be present in the files from the search
procedure. To this end the program dist can also be used:
rundist a filenum subst
coord
where coord is the name of the file with the experimental coordinates
(type spf). Since interatomic distances are strictly compared, this
structure must be energy-minimized in the same force field as was used to
create the hypothetical structures.
It is useful to
prepare a set of coordinate files in the last run of the structure generation
phase. These files must be collected for subsequent studies with other
procedures. As these procedures are usually time-consuming, the number of
coordinate files must be limited by selecting an energy range with respect to
the global minimum encountered so far. A useful script is:
select.cssr 12500 [directory]
Now all directories are scanned for CSSR files within
a window of 12.5 kJ/mol (note that the range must be given in J/mol!).
Hopefully the force field used is good enough to contain the experimental
structure(s) within the chosen range. Adding the last keyword provides a
directory into which the selected CSSR files are copied. The corresponding
script for SPF files is:
select.spf 12500 [directory]
In that directory further studies can be carried out.
In UPACK the program prep can be used, where all present files with the following
extensions can be studied in one run:
runppall.spf a subst forcefield
runppall.cssr a subst forcefield
runppall.cif a subst forcefield
Go to upack/data/etanol. Study the given connectivity file etanol.con.s, note the two charge
groups and the definition of one dihedral angle. Make the topology file by
running:
runcod etanol.con.s
runmk out.tp
Compare the resulting file out.up with etanol.tps.
To
create a coordinate file many programs exist, for instance MOLDEN. The
coordinate file cor.001 is already present in the directory. Check for trivial errors:
runpp s etanol cor.001 lj
This confirms that the coordinate file can be
optimized without problems. There are now various files named 0000…. with various extensions.
To
proceed further, the topology file has to be improved, as mentioned above. Normally this requires
careful study, using the CSD database and/or ab-initio methods. In the case of
ethanol we can use the OPLS force field, which is
supposed to work relatively well for this compound:
runcod etanol.con.a opls (note different atom labelling)
runmk out.tp opls
Here you shall note a discrepancy between out.up and etanol.tpa. The reason is that the dihedral angle C-C-O-H should not
be taken from the carbohydrate version of OPLS [9] as used
in UPACK, but rather from the standard parameter set.
For a demonstration of the random search method, go to subdirectory etanol/opls, and run:
runpa12
a etanol opls
(the actual search)
rundist a 10 etanol (clustering)
runpa3 a 13 etanol opls (first energy
minimization)
rundist a 20 etanol (second clustering)
Now we have the file pack.23 listing a set of
possible structures. To find out whether the experimental structure is present,
we have to minimize its energy in the same force field. Go to the subdirectory etanol/exp. In the database we find the experimental structures ETANOL.CIF (Pc,
Z"=2) and ETANOL01.CIF (a high-pressure form in P21/c,
Z"=1). Perform energy minimizations:
runpp a etanol ETANOL.CIF opls
or:
runpp a etanol ETANOL01.CIF opls
This creates an energy-minimized structure in two 0001.* files. Observe
that 0001.spf
equals etanol.spf or etanol01.spf, respectively.
These files are the target structures for the
structure prediction in the OPLS force field.
Search pack.23 by running:
rundist a 23 etanol
etanol.spf (search for the experimental low-pressure structure)
rundist a 23 etanol
etanol01.spf (search
for the experimental high-pressure structure)
The run was done with few trial structures (500 rather than the more
usual 10000 for Z" = 2) in one space group to reduce the computer time for
this demonstration. It is an exceptionally favourable
situation that this setting should take less than half an hour, even on an
outdated PC, to find the ethanol low-pressure structure (Z" =
Compare the resulting files with the corresponding
ones in etanol/demo. Do not worry about
slight discrepancies, the results are sensitive to processor-dependent roundoff
errors.
As seen from file distzka.pr2, the experimental low-pressure structure was found and has
Delta-E = 1 kJ/mol in this force field. The ranking is around 7. But, of
course, this refers to only one space group: a complete search would give worse
results.
The
high-pressure form ETANOL01.CIF crystallizes in P21/c with Z"=1.
Interestingly, it should also have been found in the structure generation in Pc
with Z"=2 because this is a subgroup of P21/c [4]. However, a much longer search is necessary to find this
form. This confirms that the success in finding the other ethanol
structure so quickly is a stroke of luck.
If you
wish to study details of, say, the first 10 structures, SPF-type files can be
obtained by running:
runpa3 a 23 etanol opls
rundist a 30 etanol
The input file etanol.pa3, made for this run, also codes for calculation of free
energies. It can be seen that there is some improvement in ranking.
Example: benzene
This
exemplifies a complete structure determination for a simple compound. To allow
comparison with earlier work the molecule will be kept rigid, contrary to the
normal procedure where full flexibility is ensured.
Go to upack/data/benz. Make the all-atom topology file by running:
runcod benz.con.s
runmk out.tp
Compare the resulting file out.up with benz.tps.
A special force field for benzene has been
published by Williams and Starr [10]. The coordinate file cor.001, based upon that work, is already present in the directory. Make the corresponding all-atom
topology file by running:
runcod benz.con.a opls
runmk out.tp benz
Compare the resulting file out.up with benz.tpa. The corresponding force field is benz.ff.
For the
experimental structure, go to the subdirectory benz/exp.
In the database we find the experimental polymorphs I (BENZEN01.CIF,
Pbca, Z=4) and III (BENZEN03.CIF,
a high-pressure form in P21/c, Z=2). Note the reset keyword to improve possibly unreliable
experimental hydrogen positions; this is only needed for a rigid molecule. Note
also how UPACK treats the special positions in the energy minimizations:
runpp a benz BENZEN...CIF benz
Due to the sixfold symmetry of benzene there are six equivalent possibilities
for identifying the atoms, of which you are asked to select one. In this case
the choice is irrelevant. Observe that 0001.spf
equals benz01.spf or benz03.spf,
respectively (when choice 1 is made).
The special positions make the structure prediction less straightforward, as
subgroups with the correct vale of Z must be used. For benzene several space
groups are possible, for instance P212121
for benzene I and P21 for benzene III [4].
Fortunately, for this simple compound 50 trial structures turn out to be
sufficient (rather than the more usual 5000!). This demonstration shows how 14
space groups can be treated easily with the following scripts (note that the
molecule is kept rigid throughout, so there are no intramolecular energy
contributions):
mkdirs (create 14 directories, each one containing cor.001)
rundirs1 s benz lj (the search, Lennard-Jones, followed by
clustering)
rundirs2 s benz lj (first energy minimization, Lennard-Jones,
followed by clustering)
You may
wish to try another intermolecular force field:
rundirs3 s benz pw
(second energy minimization, Buckingham, followed by clustering)
Try also:
rundirs4 a benz benz (third energy minimization, Williams-Starr,
producing SPF and CSSR files and followed by clustering)
To
investigate the clustering over all space groups, create subdirectory benz/clus
and run
there:
rundist s -23 benz
rundist s -33 benz
rundist a -43 benz
Note in allstruc.pr#
that, due to the special positions, many
structures are found in several space groups.
Search for
the experimental structures:
rundist a -43 benz
benz01.spf
rundist a -43 benz
benz03.spf
This gives
multiple solutions; if -43 is replaced by -46 there should be fewer hits.
The
experimental structures should be found in this way. However, all results are
not easy to use for survey and further work. Therefore, collect all resulting (unclustered) structures within 8 kJ/mol in subdirectory benz/collect by calling in the main directory:
select.spf 8000 collect
In subdirectory benz/collect:
runppall.spf a benz benz
(final energy minimization, Williams-Starr, producing files *prep.spf, *prep.cssr and *prep.pra)
Do a final
clustering:
cat *.prep.spf >> extra.1 (collects all
structures in extra.1)
rundist a 0 benz
Search for
the experimental structures:
rundist a 0 benz
benz01.spf (no pack
files, so number 0 is arbitrary here)
rundist a 0 benz benz03.spf
The
clusters of equivalent structures can be found in file allstruc.pr0. Both polymorphs are
found in two subgroups of the real space group.
Remove equivalent structures (or, after editing, move to another directory)
remove.1 (remove equivalent structures;
or, after editing, move to another directory)
Search for
the remaining experimental structures:
rm
extra.1 (to renew after
clustering)
cat *.prep.spf >> extra.1
rundist a 0 benz benz01.spf
rundist a 0 benz
benz03.spf
Make
a summary of all results in allres (dihedrals, here not
present, would come in alldih):
format.up
Inspect the
file allres for
the rankings of the experimental structures. The
Williams-Starr force field is excellent: the experimental structure for form I
comes out at rank 1 and form III at about 6. Compare the results with the
energies published in 1998 [11]. In that work the real space
group of each structure was investigated: due to the high symmetry of benzene,
that differs generally from the one in which it was generated.
Additional
experiments can be performed, for instance change to 30 kbar:
newcycle.spf hp (copies files to subdirectory benz/collect/hp)
Repeat in
that directory:
runppall.spf a benz benz
format.up
The new
file allres shows that the Williams-Starr
rankings for form I and form III are now about 5 and 1, respectively. So the
ranking interchanges, as it should.
Detailed
instructions for the preliminary stage
For
every coloured name a numerical value must be present in the various
files. Multiple entries should be separated by one or more spaces. A square
indicates that the corresponding entry must be given on a new line.
The space group
file upack/tops/all.spg is read by the subroutine leesr in various programs. It
must have the following entries for every space group:
naam
where naam is the space group name
nspgr is imp
where nspgr is the number of space group symmetry operations (E
excluded), is
is the space group number and imp is the importance number
for discarding structures in the clustering program dist. Space groups may
occur in different settings, with numbers usually increased by multiples of
1000.
For every
operation 1... nspgr:
msp(x) tsp(x) msp(y) tsp(y) msp(z) tsp(z)
where an equivalent position x' (in fractional
coordinates) is found from:
x' = x × msp(x) + tsp(x). Each msp is either +1 or -1 and each tsp is either 0 or 0.5, which means that space group Fdd2
is not implemented.
iref (1..12) = 0/1: disables/enables optimization of parameters:
|
cell axes: |
1: ax |
2: by |
3: cz |
|
cell angles: |
4: bx |
5: cx |
6: cy |
|
molecular centers: |
7: X |
8: Y |
9: Z |
|
molecular orientations: |
10: φ |
11: θ |
12: ψ |
itest(1...3) = 0/1 disables/enables skipping certain areas in the grid
search:
itest(1) = 1: enables
skipping of cells with ax > by
itest(2) = 1: enables skipping of cells with by
> cz
itest(3) = 1: enables skipping of cells with ax
> cz
The file upack/tops/codname.cod is needed for the program codes. It defines symbols for various
types of atoms and code numbers for terms in the force field. It has to be
prepared only once for a certain class of molecules; if desired, several files
ending with the extension .cod may be present. It must contain the following blocks
of data, separated by one blank line (=), where A1, A2, ... indicate the fixed symbols
for certain atom types and X stands for any atom type:
regel: one line of
arbitrary text
=
A1 amass mpol: atom symbol, atomic mass (a. u.), code for
polarizability (800-899).
=
A1 A2 code
for bond stretching (100-199)
=
A1 A2 A3 code
for angle bending (200-299)
X A2 X code also allowed
=
A1 code for
improper dihedral deformation (300-399). This covers all sequences A1 X X
X .
=
A1 A2 A3 A4 code for dihedral angle torsion (400-499)
X A2 A3 X
code also allowed
=
Optional: any number of sets of two lines:
A1 A2 A3... (defining one charge group)
codes for corresponding charges
(500-599).
Van
der Waals parameters are entered directly in the force field file.
Note: in case of repeated entries only the first one
is used, except for dihedral angle torsions where all relevant contributions
are added. The general "atom" type X
is only used in the absence of the exact atom
sequence, to allow the possibility of different periodicities. (This was not so
in earlier versions of UPACK.)
The molecular connectivity
file subst.con correspoonds to a certain
codes file codname.cod. It must contain the following
data:
regel: arbitrary
text
nrp: Ag, the number of atoms in the group of
molecules.
For every sequence number 1...nrp:
panm code mcg:
panm: atom name (freely
chosen, but starting with C, H, O...; e.g.C17)
code: atom type
code, corresponding with A1 , A2, ... in codname.cod (e.g.CR)
mcg: atomic charge
code as in the force field to be used. A negative mcg denotes the end of a
charge group. If only entries +1 or -1 are given, the charge code number is set
to 599, corresponding to zero charge.
inb (1...): sequence numbers of atoms (as given in the
preceding list 1...nrp) that are connected by bonds.
Repeat ad libitum on continuation lines: each line forms a chain of connected
atoms. Bonds may occur more than once. The distinction between different
molecules is simply made by the absence of connections (but note that the atoms
of each molecule must be grouped together).
ndihz: Nz, the
number of essential dihedral angles. For every dihedral 1...ndihz:
iome jome kome
ndihh: Nh, the number of unimportant
dihedrals. For every dihedral 1...ndihh:
iome jome kome
ireftr (1...3), ireftr (4...6),…..:
reference atom numbers to define the local axes system for each molecule in the
group.
For every
substance subst a topology file subst.tp# must be prepared. This can be done manually, but it is more easy to run
the program codes:
runcod subst.con codname
This program
needs two input files:
in.con (unit 18), linked to subst.con, giving the molecular
connectivity (vide infra).
in.cod (unit 19), linked to upack/tops/codname.cod, containing the force field code numbers (vide
supra)
It
produces the output file:
out.tp (unit 20), a framework for the
required topology file.
Hopefully
the contents are self-explaining. Usually some editing is needed:
Parameters may be
missing in the file codname.cod. This situation is
indicated by the warning ***** on the screen and in out.tp.
For united-atom force fields it is
necessary to fix the chirality in groups like (CH)R1R2R3
where unwanted inversion might occur. This is done by means of improper
dihedral angles like C-R1-R2-R3 which should
take values of about -35°or +35°, and so for every asymmetric atom
the choice between corresponding codes must be made.
In aromatic rings the proper
dihedrals can have the same function as improper ones. In saturated rings it
may be useful to add improper dihedral angles with weak force constants to
prevent unwanted ring inversions; for instance, improper dihedrals for the ring
atoms (of about +60° or -60°) can be used to fix one of the two possible chair
forms in six-membered rings.
The program tries to find atoms C1,
C2, C3 to define a reference axes system for the first molecule and equivalent
ones. Numbers of other reference atoms may be provided. For different molecules
(usually crystal water) the first three atoms are taken.
The torsional codes may be followed
by a multiplicity, mul. Normally this entry is
not used, but some force fields are designed to use only one of the mul possible combinations X-A-B-Y for all atoms X
and Y around two given atoms A and B. This can be implemented by setting np negative for that entry
in the force field file. UPACK will still calculate all torsions defined in the
topology file, but with the force constant divided by mul.
After
such editing, use out.tp with a suitable force field to obtain the topology file out.up:
runmk out.tp [forcefield]
Then rename out.up to subst.tp# (# = s,
a, ...).
Examples are to
be found in the directories data/glucsa, data/benz and data/etanol. It is instructive to compare the files out.up with the given topology
files. As seen for ethanol, the all-atom carbohydrate OPLS force field (vide infra) is not adequate, and manual correction was
necessary.
For the crystal
structure prediction molecular geometry files are needed for every conformation
to be studied. They are named cor.001, cor.002, ... Allowed are the spf, mdi, xyz, and cssr formats. For the
construction of these files see under Preliminaries.
The mdi format is compatible
with the GROMOS program package. It uses fixed format:
regel (a80): one line of arbitrary text
nfound (i5): the number of
following atoms. For every atom 1... nfound:
name xr
(1...3)
(10x,a5,5x,3f8.5): atom name, x, y, z (Cartesian nanometer
coordinates)
box (1...6) (free format): a, b,
c (nm), α, β, γ (degrees). This entry is omitted for a
free molecule. If used, the space group information must be given elsewhere.
The xyz format is designed
for free molecules. It is useful for interfacing with other program packages,
for example MOLDEN, where it can transfer atomic charges.
It
uses free format:
nfound: the number of
following atoms.
regel: one line of arbitrary text
For every atom 1... nfound:
name xr (1...3) [cg]: atom name, x, y,
z (Cartesian Angstrom coordinates); atomic point charge, if desired.
The
spf format
is easier to use since it is in code-directed free format:
TITL regel: one line of arbitrary text
CELL box (1...6): a, b, c
(Angstrom), α, β ,γ
(degrees). This entry is omitted for a free molecule.
SPGR spname: space group name (also
omitted for a free molecule).
ATOM name xr
(1...3):
atom name, x, y, z (one line for each atom). These lines
have fractional non-orthogonal coordinates if CELL is present, and Cartesian Angstrom
coordinates if CELL
is absent.
END (optional if there is only one coordinate set
present)
Lines
not starting with any of these code words are skipped.
The cif format should be able
to read the standard crystallographic information file. Please report errors.
The cssr format is useful for
interfacing with other program packages, for example TINKER. It is in fixed format:
box (1...3) (38x, 3f8.3): a, b,
c (Angstrom)
box (4..6) ispg spname
(21x,3f8.3,9x,i3,a20): α,
β ,γ (degrees), space
group number, space group name
nfound ispf
titel (2i4,1x,a60): number of atoms; ispf= 0: fractional
coordinates; ispf= 1: orthogonal Angstrom coordinates; first title
regel (a80): second title. Here used for
structure number and energy.
For every atom 1...nfound:
(i4,1x,a4,2x,3(f9.5,1x),8i4,1x,f7.3)
atom number, atom name, x, y, z (fractional coordinates), connectivities (max. 8), and optionally charge.
These
files are read by the procedure leesx in various programs.
Neither the names nor the order of the atoms need to correspond with the
topology, but arbitrary cell translations or symmetry operations of any atom
are not allowed. Missing H atoms are automatically generated, providing an easy
change from a united-atom force field to an all-atom force field. More than one
group of atoms may be present.
Note:
this ordering algorithm (subroutine ident) does not appear to be foolproof for large and/or highly
symmetric molecules.
As discussed
above, the force field is defined by the file upack/tops/ffname.ff. In the ethanol example we encountered simple.ff and opls.ff. As an example for the
Buckingham potential, the Williams and Starr [10] force
field benz.ff was used for benzene. More details of
these and other force fields with fixed charges, now mostly outdated as charges
should preferably be calculated for each substance individually, can be found separately elsewhere.
The force field
file upack/tops/ffname.ff is read by the subroutine leest in various programs. To
allow an easy change of force fields, the relevant information is transmitted
through code numbers as defined in the file codname.cod(vide supra). Upon
reading a force field file these code numbers are translated into actual
numerical values. The force field files must have the following structure:
regel: arbitrary text
ibuck = 0 (or -1, see below): use Lennard-Jones potential
= 1: use Buckingham potential
cutoff alphac
alpha6 hmaxc hmax6 tole tolk where
cutoff: radius for nonbonded interaction (on a charge group basis)
alphac (nm-1): Ewald-Coulomb damping by alphac × r
alpha6: same for dispersion (r -6) terms
hmaxc: Coulomb cutoff radius in reciprocal space (nm-1)
hmax6: same for dispersion (r -6) terms
tole: omit an energy term from the pair list if it contributes less than tole
tolk: same for the list of reciprocal vectors
If alphac = 0 or alpha6 = 0 the corresponding Ewald-summation is skipped.
npp [poltol schaal gamma nnn mmm nte nttt]
npp = 0: do not calculate
polarization
npp = 1: calculate
intermolecular polarization, neglecting the contribution of the induced dipoles
to the electric field.
npp > 1: maximum number
of cycles in iterative calculation of the complete polarization energy. Then
add (optionally):
poltol schaal gamma nnn mmm nte nttt
if you wish to change the default values 0.01 0.1662
0.85 3 32 1 1. See comments in subroutine nonbep for
explanation of these symbols. This option is not recommended since it consumes
extremely much computing time and because it may lead to catastrophically large
polarization energies, for which unconvincing ad-hoc solutions are introduced [12].
fact facr: factor to convert the energy
input units to kJ/mol and factor to convert distance input units to nanometers,
respectively
sc14c sc14lj iddc [epsil]
sc14c sc14lj multiply Coulomb 1...4 interactions
and Lennard-Jones 1...4 interactions, respectively
iddc [epsil]:
regulates use of a relative dielectric constant ε (default value for epsil is 1.0)
relative dielectric constant ε = epsil if iddc = 0
relative dielectric constant ε = epsil × r if iddc = 1 (note: r in Angstroms!)
Now the force field data are read, in arbitrary order and possibly mixed
with blank lines. The programs use the units nanometer for length and kJ/mol
for energy. However, in the force field file different units may be used, which
are converted by the factors fact and facr. Angles are in degrees,
but angle force constants kθ
and kξ are in
energy units rad-2. The following contributions to the force field
are implemented:
code
(100-199) cb b0 [cha]: kb , b0 in the harmonic
bond stretching potential:

Additionally, cha is an optional charge increment, to be subtracted from the
charge at the first atom and added to the charge at the second atom. Take care
to have these two atom types in the desired order in the topology file
(preferably through codname.cod). Note also that this increment does not replace the
charges described below (code 500-599) but is added to them.
code
(200-299) ct t0: kθ
, θ0 in the
harmonic angle bending potential:
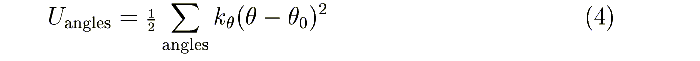
code (300-399) cq q0: kξ
, ξ0 in the harmonic improper dihedral angle potential:
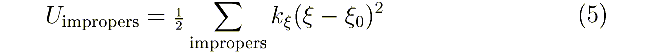
These terms are necessary to define the chirality
around united-atom CH groups and to prevent the deformation of rings and planar
groups.
code (400-499) cp np [cp np ...]: Vn,n,
... in the cosine torsional potential:
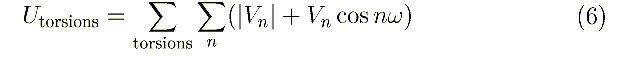
Some force fields are designed to use only one of the
possible combinations X-A-B-Y for all atoms X and Y around two given atoms A
and B. This can be implemented by setting np
negative for that entry in the force field
file. UPACK will still calculate all torsions defined in the topology file, but
with cp divided
by the multiplicity (see also the topology file).
code (500-599) cg: qi
(elementary charge units) in the Coulomb interaction:

where ε
is the relative dielectric constant, and qi is multiplied
internally by the factor 11.787 needed to obtain the energy in kJ/mol. The
1...4 interactions are multiplied by sc14c.
The van der
Waals parameters for nonbonded interactions are treated differently since they are
based strictly on atom types rather than on individual atoms. The cards start
with two atom types A1 A2,
followed by numerical data. Entries for the combination of two equal atoms must
be present; if other interaction parameters are absent (or are given the value
-1), they are found from a combination rule. For the combination of atom types i and j this rule is generally the geometric
average:
![]()
but sometimes
the arithmetic average is used:
![]()
Intramolecular 1...2 and 1...3 interactions are always excluded. For the
Lennard-Jones interaction it is possible to specify separate parameters for 1...4
interactions. Missing (or negative) parameters for the combination of two equal
atoms are taken from the corresponding general parameters. For missing mixed
1...4 parameters the rule is:
sc14lj < 0: set equal to the corresponding general parameter
sc14lj > 0: use the combination rule for 1...4 parameters
Subsequently, all 1...4 interactions are multiplied
by |sc14lj|.
The following input formats are used:
If ibuck= 0: A1 A2 C12 C6 [C12(1...4)
C6(1...4)]
These
are the parameters for the Lennard-Jones interaction:
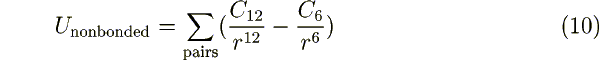
The
geometric combination rule applies.
If ibuck = -1: A1 A2 E R [E(1...4)
R(1...4)]
As
above, but now C12 and C6 are found
indirectly:
![]()
The
geometric combination rule applies for E and the arithmetic rule for R.
This precludes the use of Ewald summation for the attractive terms.
If ibuck = 1: A1 A2 A B C
These
are the parameters for the Buckingham interaction:
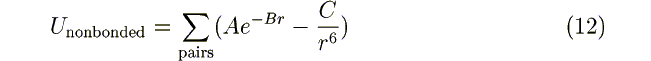
There
are no special 1...4 interaction terms in this case. The geometric combination
rule applies for A and C, and the arithmetic rule for B.
code
(800-899) alfa: αi (volume units), the polarizability of
atom i in:
![]()
where Ei
is the electric field strength at that atom.
It is possible
to introduce charges on "virtual atoms", i.e. on sites which are
defined in terms of three real atoms a, b, c, as follows:
![]()
where
p and q are dimensionless fractions and s is in A-1.
For each separate molecule (01, 02, 03, ...) these
sites must be defined in the files mol01.def, mol02.def, ....., containing for each virtual atom:
a b p [c q [s]]
where
a, b, c are atom numbers as defined for that molecule
(which is different from the topology file for all molecules after the first
one!). Absence of such a file means that for the corresponding molecule only
the normal atomic sites are considered. So this feature can also be used to
simply supersede the charges defined in the force field file. Note that the
atom numbers start at number 1 for each separate molecule.
If this file is
present, there must also be the file chadd.xyz, containing the
charges. For each distinct molecule there must be a block of data in XYZ
format:
nxyz: the number of atoms
(real + virtual)
titel: comment. Then for
every atom 1... nxyz:
atom name (X for virtual atoms),
various data (not used) with last entry: charge
Apart
from the virtual atoms, the atom order must be the same as in the topology
file. This feature can also be used in the absence of files mol#.def. Then it simply
overrules the charges from the topology file; for this purpose the file chadd.xyz may also be in the cssr format.
Note: although this feature looked
promising, it turned out not to give better results than the usual atomic point
charges.
Detailed instructions
for the search stage
In
the following the standard file setup will be assumed. The space group file is
always taken from upack/tops. Topology files and force field
files are first sought in the directory where the execute script is called. If
they are not found there, they are sought in a reference directory (upack/data/subst and upack/tops, respectively).
All programs
have code-directed input. After each code word one or more numerical entries (separated by one or more spaces)
may be required or optional. If a certain code word is not specified, the
parameters keep their default values which are given between parentheses {...}.
The same goes for unspecified optional parameters (indicated by [...]). Code
words that are unknown, or preceded by one or more spaces, are disregarded.
A number of
code words occurs in several programs. Some are discussed here, and will be
mentioned in the individual input descriptions without repeated explanation.
cutoff cutoff [alphac alpha6 hmaxc hmax6 tole tolk]
Overrides the cutoff value given in the force field
file, and possibly also the settings for Ewald summation .
epsil epsil
Overrides the dielectric constant value given in the
force field file.
inner inner
A surface charge on polar crystals causes a
shape-dependent energy term. Normally it is assumed that this term is
compensated by external charges; this corresponds to omitting the h=0
term in Ewald summation [13]. If the same situation is to be
simulated without Ewald summation, a term 2πp2 / (3 V)
must be added. In UPACK this is done if inner
> 0; if alphac > 0 inner is set to zero. In lattice vibrational calculations, the
contribution of this term to the second derivatives is omitted unless inner = 2.
pol npp
[poltol schaal
gamma nnn mmm nte nttt]
Include polarization, possibly overriding
the default values given in the force field file.
sd# ncy#sd tol#sd stp#sd
nre#sd shm#sd
Specifies steepest descents energy minimization in
various stages (#) of a program: the minimization is stopped after ncy#sd cycles or earlier if the energy decreases less than tol#sd. The initial step size is determined by stp#sd. A new pair list is made after nre#sd cycles or if any parameter changes more than shm#sd. The steepest descents minimization is usually followed
by:
cg# ncy#cg
tol#cg stp#cg dep#cg shm#cg
Specifies conjugate gradients energy minimization in
various stages (#) of a program: the minimization is stopped after ncy#cg cycles, or earlier if
the root mean square value of the energy derivatives is less than tol#cg and a recalculation with a new pair list does not change
the energy more than dep#cg. The initial step size is determined by stp#cg. A restart with a new pair list is made if any parameter
changes more than shm#cg.
files ifils
ifilm ifild ifilc
Produce output files if these parameters are larger
than 0:
ifils = 1: format spf.
ifilm = 1: format mdi.
ifilm = 2: format xyz.
ifild = 1: format cif.
ifild = 2: format fdat.
ifilc = 1: format cssr.
ifilc = 2: format cssr, the charges
are written out too.
This program
generates structures with any number of groups of molecules. It performs the
same action as the combined grid search programs pack1 and pack2, but in quite a different way.
Rigid molecules are placed with random positions and random orientations in a
cell with randomly chosen crystallographic parameters. Dihedral angles may be
set to random values too. The resulting structures are optimized with respect
to energy, assuming either rigid or fully non-rigid molecules. An arbitrary
number of conformations can be studied consecutively.
It is possible to make use of partial knowledge by
limiting the search range for the cell axes and cell angles. It is also
possible to take fixed starting values for known positions and orientations of
molecules, as well as for dihedral angles.
Execute
script:
runpa12 s/a/... subst ffname
Input
files:
in.tp (unit 20), molecular
topology, linked to upack/data/subst/subst.tp#
in.spg (unit 11), space group, linked to upack/tops/all.spg
in.ff (unit 12), force field, linked to upack/tops/ffname.ff
in.pa (unit 13),
code-directed input, linked to subst.pa12
cor.001, cor.002, cor.003, ... (unit 14),
coordinates for various conformations.
mol#.def (unit 48,
optional): a file defining additional charge sites on virtual atoms.
chadd.xyz (unit 48, optional): a file containing charges to
supersede the values from the topology file, and possibly also defining the
additional charges on virtual atoms.
The number of
distinct groups of molecules (G) is called ngr. This number must
certainly not be larger than 4 to avoid excessively long and usually fruitless
computations. Each energy calculation is discontinued immediately if any
repulsion term exceeds the value ebig. This will occur in the
majority of cases. If not, ncysd cycles of steepest
descents and ncycg cycles of conjugate gradients
energy minimization follow.
The coordinate
file(s) must contain the atoms in exactly the same order as the topology file.
If ngr is larger than one, the coordinates can either be specified for each
group individually or for a number of groups. In the latter case ngr
must be a
multiple of that number, and the given coordinates are copied as many times as
necessary.
If one decides
to stop the calculation before the originally desired number of structures is
found, a "clean" stop can be forced by creating a file called STOP. This will halt the program as soon
as the next multiple of 1000 accepted tries is reached. If the file STOP contains an integer number, the calculations
are stopped after that number of structures, thus replacing the parameter nrand.
The values of ebig and emax1 are determined automatically, increasing
with the number of atoms, unless set by the corresponding code words.
Output
files:
out.pri (unit 16), general results, linked to pack.pr1
pack.10 (unit 10),
structure parameters
pack.19 (unit 19),
coordinates
Input
file
The input data
discussed above can be manipulated by cards in file in.pa.
ngr ngr{1}
The number of groups of molecules, G.
rand nrand{5000}
The search continues until nrand random
structures are found (for every conformation cor.001, cor.002, cor.003, ...).
spgr naamsp
The space group name (upper or lower case, slashes
may be omitted). This card must always be present, except when the name of the
directory is the same as that of the space group.
emax emax1 [emax2] {40 (or higher) 10000}
Structures with energy higher than emax1 (with respect to
the lowest energy found at that time) or higher than the absolute value emax2 are not included
in the output file.
axes amin amax bmin bmax
cmin cmax {0.4 2.5 0.4 2.5 0.4
2.5}
The cell axes are scanned: ax = amin...amax, by =
bmin...bmax, cz = cmin... cmax. This option disables autmax.
autmax autmax {3.7}
For large cells the axes are increased to autmax × sqrt (cell volume). This option is disabled if autmax is negative or if axes is set.
angles bxmin
bxmax cxmin cxmax cymin cymax {0 1 0 1 0 1}
The cell angles, represented by the projections of
the cell axes on the Cartesian axes, are also varied:
bx = (bxmin...bxmax) × ax, cx = (cxmin...cxmax) × ax, cy = (cymincymax) × by.
grvfix xxfix yyfix zzfix (1...Mg × Gi )
{1000...1000}
Fixed starting centers of gravity for each molecule
(but random for each value > 999)
eulfix phifix thefix psifix (1...Mg × Gi )
{1000...1000}
Fixed starting Euler angles for each molecule (but
random for each value > 999)
hydfix hydfix ((1...Nz+Nh),1...Gi
)) {1000...1000}
The starting value of each dihedral angle is set to
the corresponding value of hydfix, except:
if hydfix > 999, random values are assigned to dihedral
angles
if hydfix < -999, the dihedral
angles from the input geometry are retained
celopt icopt {1}
Cell axes and angles are kept fixed if icopt = 0.
sd ncysd tolsd stpsd nresd shmsd {100
1 0.001 10 1}
cg ncycg tolcg stpcg
depcg shmcg {1000 1 0.01 1 0.05}
ebig ebig{2000 … 80000}
dexp dexp {-100} [volcor {1.10}]
dexp > 0: The experimental density dexp (in gram cm-3) is used to find ax.
dexp = 0: No such restriction on ax. However,
this is usually very inefficient since much time is wasted on unproductive
starting structures.
dexp < 0: The density is estimated from - dexp random structures (for
each conformation) before starting the calculations. Its value is found from
the smallest volume encountered.
volcor: The smallest volume encountered is multiplied by volcor to facilitate finding
new structures.
inner inner {1}
seed iseed {281197}
The integer iseed initializes the
random number sequence.
append iseed
If this keyword is present, the nrand random structures are
appended to the results of a previous search (pack.10 and pack.19). It is necessary to redefine seed iseed here to prevent just
repeating the previous calculation! If the previous calculation was stopped
uncleanly, take care that pack.10 and pack.19 end with the same entry.
coul icoul {1}
If icoul = 0 no Coulomb interactions are calculated.
chadd ichad{0}
ichad = 0: charges from topology file
ichad = 1: charges from file chadd.xyz, but
only for real atoms
ichad = 2: charges from file chadd.xyz, also
for virtual atoms.
intra intra {1}
intra = 0/1: disable/enable
the calculation of intramolecular nonbonded interactions
flex iflex {1}
iflex = 0: rigid molecules, only intermolecular nonbonded
interactions. This option implies intra = 0.
iflex = 1: fully flexible molecules
pres pres {0}
The pressure in atmospheres.
print iprint {0}
Minimal output on the screen is given for iprint =
test dtest {0}
If dtest > 0, the analytical energy derivatives are compared
with numerically calculated values using parameter shifts dtest (dtest = 0.00001 is usually a good value).
conn icn {0}
If the subroutine leesx fails, it
calls the subroutine connect to try and repair the situation. The geometry is compared
with the topology in order to assign the correct atom numbers to the
coordinates. There may be more solutions; usually they are identical but the
program must be guided to select the correct solution number icn. It will ask you to enter that number manually. However,
usually the program is called from a script and this will not be possible. Then
it is necessary to set conn with the desired solution number icn.
cutoff cutoff [alphac alpha6 hmaxc hmax6 tole tolk]
Overrides the default values from the force field.
However, the values from simple-lj.ff, opls.ff or unitat3.ff (0.8 3 3 2 2 0.02 0.02) are recommended: Ewald summation
with a still fairly small cutoff radius and elimination of quite a lot of terms
in the pair list.
epsil epsil
Overrides the dielectric constant given in the force
field file.
stc stcin(1...4) {1 1 1 1}
Steepest descents weights for cell axes, cell axis
projections, and atomic coordinates, respectively.
knot
This codeword enables the search for knotted
structures.
Output
files
The file pack.10 contains first data concerning
space group symmetry, torsional degrees of freedom and atomic coordinates of
the free molecule. For explanation of the symbols see the space group and
topology files. The following lines occur:
- ispgr
icode
Space group number and sequence code. The sequence
code is -1 to indicate that the file is generated by program pack12.
- ndihz ndihh:
- iome jome kome
lome
- nrp ireftr (1...3): Ag, followed by the three
numbers of the reference atoms.
Then for every atom i =
1...nrp:
- i panm iac
xr (1...3),
where panm is the atom name
corresponding to the topology file, iac is the sequence number
of the atom type code, and xr gives the Cartesian
coordinates xi, yi,
zi in nm for the first geometry file (cor.001).
- cel (1...12): two
lines with starting values for the parameters
- iref (1...12): one line to
control the optimization of 12 parameters
After these initial
data, each line characterizes a certain structure:
code1: N1,
the sequence number of this structure in the random series (after the density
test but before the energy test)
code2: N2,
the sequence number in this file.
This is kept as identification number in all
subsequent calculations.
epot: the energy (kJ/mol)
par (1...): ax,
by, cz, bx,
cx, cy (pm) unless constrained by symmetry
(i.e, the corresponding iref must
not be zero).
en1 vol:
the energy value at the initial calculation, and the molecular volume (both
multiplied by 1000 to obtain an integer value)
tau (1...): the
values of the dihedral angles
1 (the multiplicity, for later reference)
Clustering by dist
yields the
file
pack.13 which
has the same structure. Only code1 now gives the sequence number in the clustered file (code2 still refers to N2)
and two new items are added:
zflag: the number of independent molecules (Z'')
imult: the cumulative number of structures that have been clustered
together for this entry
The file pack.19 contains Cartesian
atomic coordinates for every structure:
- icod2 box (1...6): N2,
ax, by, cz,
bx, cx, cy
- x (1...3Ag): x1, y1,
z1, x2, y2, z2,
...
This program performs
the energy minimizations in subsequent stages of the crystal structure
prediction. Either rigid models or fully flexible ones can be treated.
Execute
script:
runpa3 s/a/... filenum subst
ffname
To illustrate the
use of the file number, we take filenum = 13 (the first
instance where pack3
is usually called). As an alternative example between {...} we take filenum= 42; this should make
the general principle clear.
Input
files:
in.tp (unit 20), molecular
topology, linked to upack/data/subst/subst.tp#
in.spg (unit 11), space group, linked to upack/tops/all.spg
in.ff (unit 12), force field, linked to upack/tops/ffname.ff
in.pa (unit 13),
code-directed input, linked to subst.pa2{5}
in.par (unit 21), structure parameters, linked to pack.13{42}
in.cor (unit 29), coordinates, linked to pack.19{49}
in.add (unit 14), optional set of additional coordinates, linked
to file extra
mol#.def (unit 48,
optional): a file defining additial charge
sites on virtual atoms.
chadd.xyz (unit 48, optional): a file containing charges to superside the values from the topology file, and possibly
also defining the additial charges on virtual atoms.
Output
files:
out.pri (unit 16), general results, linked to pack.pr2{5}
out.par (unit 30), generated structure parameters, linked topack.20{50}
out.cor (unit 39), generated coordinates, linked to pack.29{59}
For every structure (identified with a five-digit
number #) there is the possibility for coordinate output in four formats (unit
17), linked to #.spf, #.xyz, #.mdi, #.dat, #.cssr.
Additional possible output from molecular dynamics:
#.en (unit.15),
energy snapshots
#.co (unit.17),
coordinate snapshots
#.ve (unit.18),
velocity snapshots
#.xvo (unit.19), final
coordinates and velocities
If one decides
to stop the calculation before the originally desired number of structures is
found, a "clean" stop can be forced by creating a file called STOP.
The program
automatically detects the number of molecular groups from the number of atoms
on the coordinate file. It is also possible to read an (additional) hand-made
file in.add with cell parameters and atom coordinates.
It is possible
to expand the geometry to independent molecules in space group P1. But
the energy still refers to one molecular group. The objective is to see whether
the symmetry is destroyed when it is no longer enforced; if so, the structure
is in fact unstable. In this situation a significant change in energy occurs,
and the resulting symmetry-averaged coordinates do not make sense.
The same
objective can be reached by performing a primitive MD-run before (or instead
of) the energy minimization. Here too one has the choice whether or not to
enforce the space group symmetry. It is obviously better not to do so; in fact,
to make the simulation less primitive one should consider more than one unit
cell. That option is available in the program crysmd.
It is possible
to calculate the lattice vibrations and the associated thermodynamical
quantities. Here the occurrence of imaginary frequencies is an indication that
the crystal structure is not a stable one.
The value of emax1 is determined automatically, increasing with
the number of atoms, unless set by the corresponding code word.
Subsequent
runs
To continue the
energy minimization, subsequent runs can be done after clustering. For
instance:
rundist a 20 subst
creates pack.23, and
runpa3 a 23 subst ffname
uses the parameter file pack.23, together with
input file subst.pa3 and coordinate file pack.29, to produce results in
pack.pr3,
pack.30 and
pack.39. This procedure can be repeated at
will up to pack.pr9.
Input
file
The options
mentioned above can be manipulated by cards in file in.pa:
expand expand
{0}
If this code word is present, the coordinates are
expanded to P1. Random position disturbances (with maximum expand) can be added to
move the system slightly away from a possible saddle point; a reasonable value
would be 0.0005.
maxinp maxinp {5000}
The maximum number of input structures to be read
from in.par. To select certain structures from this file, set maxinp < 0 and then give on the same line any desired quantity
of sequence numbers N2. If no sequence numbers are given, the
program disregards the information from in.par and
operates on all entries from in.cor.
emax emax1 [emax2] {30 10000}
The highest allowed energy: emax1 is relative to the
first entry on the input file in.par, emax2 is the absolute
value.
celopt icopt {1}
Cell axes and angles are kept fixed if icopt = 0.
sd ncysd tolsd stpsd nresd shmsd {100
0.01 0.0001 5 0.02}
cg ncycg tolcg stpcg
depcg shmcg {5000 0.0005 0.005 0.3 0 .02}
These values ensure good convergence, but this will
cost a lot of computer time. If the number of structures is large, an
intermediate calculation with fewer cycles and a larger tolerance will be more
efficient. However, equivalent structures may then not be recognized in the
clustering procedure.
extra [ncycsd tolsd
ncyccg tolcg]
If a file in.add is present to
provide additional structures, new values for the minimization parameters can
be given here. The format of this file may be mdi or spf; in the latter case the data for the different molecules
must be separated by an END card. If the file title starts with a number that has not
been used before, that number identifies the structure. Otherwise a new number
is assigned, starting with 90001.
inner inner {2}
coul icoul {1}
If icoul = 0 no Coulomb interactions are calculated.
chadd ichad{0}
ichad = 0: charges from topology file
ichad = 1: charges from file chadd.xyz, but
only for real atoms
ichad = 2: charges from file chadd.xyz, also
for virtual atoms.
intra intra {1}
intra = 0/1: disable/enable the calculation of
intramolecular nonbonded interactions
flex iflex {1}
iflex = 0: rigid molecules, only intermolecular nonbonded
interactions. This option implies intra = 0.
iflex = 1: fully flexible molecules
pres pres {0}
The pressure in atmospheres.
iprint {0}
Minimal output on the screen is given for iprint=
test dtest {0}
If dtest > 0, the analytical energy derivatives are compared
with numerically calculated values using parameter shifts dtest (dtest = 0.00001 is usually a good value).
cutoff cutoff [alphac alpha6 hmaxc hmax6 tole tolk]
The default values from most Lennard-Jones type force
fields are chosen for speed rather than accuracy. For instance, the
values 0.8 3 3 2 2 0.02 0.02 give Ewald summation with a fairly small cutoff
radius and elimination of quite a lot of terms in the pair list. This is a
reasonable compromise between speed and precision. But note that some
calculations may not converge, or even give absurd results. Redo these (using
the maxinp -1 ... option to save time) with, for instance, cutoff 1.2 3 3 2 2 0 0.
Especially in the last stages of the analysis it is better to have tole = tolk = 0, and to experiment
with increasing the other parameters.
epsil epsil
Overrides the dielectric constant value given in the
force field file.
pol npp [poltol schaal gamma nnn mmm nte nttt]
files ifils
ifilm ifild ifilc {0 0 0 0}
stc stcin(1...3) {1 1 1}
Steepest descents weights for cell axes, cell axis
projections, and atomic coordinates, respectively.
knot
This codeword enables the search for knotted
structures.
If
molecular dynamics calculations are desired:
mdrun nstlim {0} [nrun {1}]
Do molecular dynamics. This is done by subroutine runmd, which has been taken from the GROMOS program package
and adapted for use in crystal simulations. See ref. [14]
for background. Time units are ps.
The card mdrun must give nstlim, the number of MD steps. Optionally the number of runs (nrun) may be specified. The following cards may also be used:
mdcut cutoff [alphac alpha6 hmaxc hmax6
tole tolk]
Only needed if different from the values used for the
energy minimization
mdtsc ntt tempi tautp {1 0 0.10}
Controls temperature scaling:
ntt = 0: disable temperature scaling
tempi: if tempi > 0 the
initial velocities are taken from a Maxwellian distribution with T = tempi
tautp: relaxation time for temperature scaling
mdtemp temp0 {298}
temp0: reference
temperature for velocity scaling
mdpsc ntp comp taup {4 0.00076 5.0}
Controls pressure scaling:
ntp = 0: no pressure scaling
ntp = 1: isotropic pressure scaling
ntp = 2: orthorhombic pressure scaling
ntp = 3: monoclinic pressure scaling
ntp = 4: triclinic pressure scaling
comp:
compressibility (Gromos units), essentially only a
scaling factor
taup: relaxation time for pressure scaling
mdpres pres0
pres0: reference pressure (atm)
Only needed if different from the pressure used in
the energy minimization
mdshak ntc {3} [tol {0.00001}]
ntc = 1: disable the use of bond length constraints by the shake procedure.
tol: the geometric tolerance for that procedure
mddt dt
{0.002}
dt: the time step
in picoseconds
mdnscm nscm {500}
Stop translation of the center of mass after every nscm steps
mdnsnb nsnb {10}
Make a new pair list after every nsnb steps
mdpri ntpr ntwx ntwv ntwe ntpw {0 0 0 0 2}
Write output to screen after every ntpr steps (the quantity of output being determined by iprint).
If ntwx > 0, write
coordinates to #.co (unit 17) after every ntwx steps.
If ntwv > 0, write
velocities to #.ve (unit 18) after every ntwv steps.
If ntwe > 0, write various
results to #.en(unit 15) after every ntwe steps.
If ntpw = 0, the coordinate
and velocity files are unformatted.
If
lattice vibration calculations are desired:
latvib [nq ...] {0}
If this keyword is present, lattice vibration
calculations are performed. It is advisable to do energy minimization first.
The calculations are first done for q = 0, and subsequently for nq vectors in q-space.
Possible modes of input:
latvib nq
Calculations are done for nq randomly chosen q-vectors.
latvib n1 n2 n3
Calculations are done for nq = n1 × n2 × n3 q-vectors,
filling a regular grid in q-space.
latvib f1 f2 f3 nq
Calculations are done for the vectors defined by f1, f2, f3 divided in nq equal parts.
vibamp
If this card is present, eigenfunctions are
calculated in order to find the vibrational amplitudes.
temp vtemp(1....) {298, undefined ...}
The thermodynamical quantities (and the vibrational
amplitudes, if requested) are calculated at temperatures vtemp(1), vtemp(2), vtemp(3), ...
cutlv cutlv tollv {1.5, 0.0}
There is no Ewald summation possible, so one must be
able to choose a larger cutoff (cutlv) than in
the other calculations. Matrix elements smaller than tollv are
disregarded. Note that here the use of charge groups with appreciable net
charge may lead to inaccuracies.
Output
files
The file pack.20 contains first data concerning
space group symmetry, torsional degrees of freedom and atomic coordinates of
the free molecule. This is quite similar to the output from pack12, but note that now icode is equal to the
sequence number of the call of pack3. After these initial data, each line again
characterizes a certain structure:
code1: N1,
the sequence number in this file
code2: N2,
the sequence number from pack12. This is kept as identification number in all subsequent
calculations.
epot: the energy (kJ/mol)
par (1...): ax,
by, cz, bx,
cx, cy, X, Y, Z,
φ, θ, ψ, unless constrained by symmetry (i.e, the corresponding iref must not be
zero). Lengths in pm, angles in degrees multiplied by 10.
omega (1...Nh+Nz): torsional
angles (degrees)
imult: the number of structures that have been clustered
together for this entry in previous runs.
If more than
one independent molecule is present, only the cell parameters are given (i.e.,
centers of gravity, Euler angles and torsional angles are not included).
The file pack.29 contains Cartesian
atomic coordinates, in exactly the same format as pack.19.
The molecular
dynamics output file #.en
contains for every selected time step:
ener (1...46):
1...10: energy components: total, kinetic, potential;
ax, by, cz,
bx, cx, cy, volume
11...15: energy components: bonds, angles, impropers, torsions, nonbonded
15...20: not yet assigned
21...30: nine pressure components, total pressure
31...40: nine virial components, total virial
41...46: a, b, c (nm), α
,β, γ (degrees)
This program
compares possibly equivalent crystal structures on the basis of a comparison of
interatomic distances. It can be used to eliminate or collect equivalent
structures ("clustering mode") or to search for the presence of a
certain structure (usually the experimental one; "search mode").
Fairly well-refined structures are required: if the geometrical differences
become too large, the structural equivalency may be no longer recognized.
Execute
script: rundist u/a [-]filenum subst [coord]
Input
files:
in.tp (unit 20), molecular
topology, linked to upack/data/subst/subst.tp#
in.spg (unit 11), space group, linked to upack/tops/all.spg.
in.dcl (unit 13; optional), code-directed input, linked to:
- clustering mode:
subst.cl
- search mode: subst.zk
parameter file(s) (fort.201, fort.202, fort.203, ...): type pack.20.
coordinate file(s) (fort.101, fort.102, fort.103, ...): type pack.29.
For instance, if filenum = 41 then fort.201 is linked to pack.41 and fort.101 is linked to pack.49.
extra.1, extra.2 ,... (unit 14,
optional): additional structures in continuous
spf-type files.
in.exp (unit 15, optional): an spf-type file containing
one reference structure for search mode, linked to the file coord.
If filenum = -41, all
corresponding files in directories at the same level are linked. This means
that the call should normally be done in a subdirectory of subst (e. g. subst/clus).
Output
files:
out.pri (unit 16), linked to distcl#.pr# or distzk#.pr#.
In clustering mode also:
- new parameter file(s) (fort.301, fort.302, fort.303, ...): type pack.20.
- a combined list of surviving structures: cluslist, linked to cluslist.pr#.
- a list of clusters of all structures: allstruc, linked to allstruc.pr#.
- for the continuous spf-type file:
file(s) remove.# indicating the removed structures and file(s) select.# containing the
remained structures. A file remove.# can be directly called to remove the indicated structures,
or edited to move them to somewhere else.
Two
structures are considered equivalent if the radial distribution curves for each
atom in the asymmetric unit are the same. This is even more specific than
comparing powder diffraction diagrams, since not only the distances but also
the atom types must correspond. This type is normally equal to the atom type, but it can be set
more specific than just, say, "hydroxyl O": in glycerol the two end
oxygen atoms are equivalent, but the middle one is different.
The structures
to be compared are characterized by their space group symmetry, but there is no
necessity that this space group is the same as required by crystallographic
conventions. For instance, a P21/c structure could
also have been generated in P21 with two quasi-independent
molecules or even (with cell doubling) in P1 with four. So the numbers
of molecules in the various structures to be compared may differ. The program
first determines for every structure the number of crystallographically
independent molecules (Z'') before starting the actual comparison of
structures. There, of course, only structures with the same Z'' can be really
equivalent. The determination of the actual space group of these structures is
quite another matter, for which programs like PLATON [5]
may be employed.
Structural
information
The program
tries first to read cell parameters and atomic coordinates from a set of pack-type coordinate files,
one for each space group to be considered. They must be assigned to files fort.101, fort.102, ..., and the
corresponding pack-type
parameter files (fort.201,
fort.202,
...) must be present to select the desired structures, and to carry information
on space groups and atom types. These files have the standard UPACK format, as
described under pack3.
In the script file rundist the second parameter gives the
number of the file to be considered (e.g. 30 for the file pack.30); if this number is
given as -30 all pack.30 files present in directories at the same level (usually a directory for
every space group) will also be linked.
After that, the
program tries to find additional structures from file(s) extra.#, which contain an
arbitrary number of spf-type entries. Each entry must contain at least the
cards:
CELL box (1...6): a, b,
c (Angstrom), α, β, γ (degrees)
and cards with fractional non-orthogonal coordinates:
ATOM panm x (1...3): atom name,
x, y, z (one line for each atom). The atoms must be
ordered as in the topology file, and it must be possible to form molecules from
their coordinates without using symmetry operations.
The information for every structure is concluded by
the card:
END
Obviously, at
least either this file or one pair of pack-type files must be present. Optional information can
be given by a title card containing one line of arbitrary text:
TITL regel
and the energy can be transmitted by a card:
ENER ener
This is used for selection of structures to be
compared; in the absence of this card no such selection is done for this entry.
Symmetry
information
In each pack-type file the space
group number is given on the first line. In the additional file(s) extra.# the space group information
is presented for each entry separately, either by a space group name, e.g.:
SPGR P21/m
or by detailing a set of symmetry operations
separated by commas:
SYMM -x, y+1/2, -z
SYMM x, -y+1/2, z
where the redundant card
SYMM -x, -y, -z
is optional. If both types of cards are present, the SYMM information is
used.
Lines not
starting with any of the described code words are skipped. When only symm cards are used to
introduce space group information, the file in.spg is not used. In that
case the parameter filenum is a dummy argument, and all space
groups can be defined. But if the space group of any entry is defined
otherwise, the space group file in.spg is needed and only
triclinic, monoclinic and orthorhombic systems can be handled.
Distance
selection
It may be
necessary or desirable to modify the standard information on atom types as
given in the files discussed above for one or more of the following reasons:
- Normally only intermolecular distances are
considered. So, if the list of atoms contains distinct molecules, each last
atom of a molecule must be marked.
- Atoms may have the same type (vide
supra).
- The coordinates of some atoms, for instance hydrogen
atoms bound to carbon, carry hardly any significant information. Deleting such
atoms can lead to a substantial increase in speed, and also eliminate some
structure differences (desirable or not!).
The specification of this information is done through
an atom card
(vide infra) in the file in.dcl.
Mode
selection.
In the standard
mode of operation ("clustering") the program removes equivalent
structures. Output, sorted to energy, is written to new pack files with the number increased by
three: for instance, pack.31 is clustered to pack.34.
Alternatively,
a file in.exp may be present which defines a reference structure
(in the spf format described above). In this case, the program switches to the
"search" mode where each structure of the list(s) is only compared
with the reference structure. If in.exp is not present, the
script looks for it in directory subst/exp.
Details of the
calculation can be manipulated by cards in file in.dcl:
cutoff cutoff
{0.6}
The cutoff distance (nm) for the radial distribution
curves
margin margin {0}
The number of contacts per molecule that are allowed
not to be found
sym isymm {1}
isymm = 0: the space group is changed to P1. This
overrules the space group information given on the pack-files.
isymm = 1: space group information is used.
dener dener {1.0}
The comparison between two structures is skipped if
their energies differ by more than dener. This does not apply
to spf-type entries where no energies are available.
Also it applies only to the clustering mode.
rvol rvol {0.01}
The comparison between two structures is skipped if
the ratio between their molecular volumes deviates from 1 by more than rvol. This applies only to the clustering mode.
tol tol {0.005 in clustering mode,
Maximum allowable deviation (nm) between two
distances that are being compared.
print iprint {0}
Regulates the quantity of output: larger iprint gives more output.
import import(1)
import(2) (...) {1, 2, 3, 4, ...}
Determines importance of the lists as read by the program. There is a default
list determined by entries in the space group file all.spg, to allow discarding structures
according to space group multiplicity. Numbers for the most common space groups
are, for instance:
1
3
7
8 10 11
17 22
23
25 31 45 46
P1, P-1, Pc,
P21, Cc, C2, P21/c, Pca21, Pna21, P212121, C2/c, Pbcn, Pbca
These numbers are increased with about 950, with lower increases for subsequent
sets of space groups, and extra lowered for cases with higher values of G.
This order of importance can be changed by using the import
card.
select isel {1}
Rules details of the clustering:
isel = 0: discard the structure with lowest energy.
isel = 1: discard the structure from the list with the lowest import number (lowest
number within one list).
isel = 2: discard the structure from the list with the highest import number (highest
number within one list).
intra intri(1) intrj(1) intri(2) intrj(2) (...)
Consider also
intramolecular contacts between atom pairs numbered intri and intrj. These pairs should be
chosen to discriminate between several possible conformations of flexible
molecules; it is a probably superfluous safeguard. In the absence of this card
all intramolecular distances are omitted.
emax emax {very big}
The highest allowed energy: structures with energy
greater than emax are skipped in the clustering.
mult
This card, if present, produces the file multiples where only the
equivalent structures are indicated.
atom
This card, if present, must be the last one in the
file in.dcl and must be followed by nrp cards (one for each
atom) containing:
(-)k: a sequence number (1...
nrp); a negative k denotes the last atom
of a molecule.
panm: the atom name
type: an integer
number to indicate the atom type. These numbers can be arbitrarily assigned
between 0 and 99. If type = 0 the corresponding atom is skipped.
In the absence of an atom card, the program takes the types from the topology file in.tp. Lacking that, it
searches for the pack-type file fort.201 and sets the type equal to the type found there. If that
file is not present either, then all atoms are given a different type. Note that in the
last two cases all atoms are considered to belong to one molecule, and then for
cases with Z''>1 an atom card is necessary.
This program
calculates and minimizes the energy of one or more single (crystal) structures,
thus optimizing their geometries. The input data are obtained from an spf, mdi, cif or cssr type coordinate file. It
is possible to expand the geometry to independent molecules in space group P1
or to consider more than one independent molecule in the asymmetric unit.
Lattice dynamical calculations are also possible.
The space group
is determined by the coordinate file, where a space group name may be
specified. If this information is absent, the space group name is taken from
the code-directed input file in.pp. In case of conflict
the coordinate file has priority. If still no space group name is available,
space group P1 is assumed. The symmetry operators in the coordinate file
are combined with those of the space group. If no cell data are given, a free
molecule is assumed. The number of independent groups of molecules (Gi)
is determined from the coordinate file. Neither the names nor the order of the
atoms need to correspond with the topology, and arbitrary cell translations of
any atom are allowed. If hydrogen atoms are missing in positions that can be
guessed easily, the program is able to provide these missing coordinates.
Structures with
molecules on special positions or with symmetries higher than orthorhombic are
treated in a roundabout way. In the former case the symmetry operators which
cause atoms to coincide with themselves or with others are eliminated; in the
latter case the molecules which are created by operators that cannot be handled
by the program are replaced by "independent" ones. Thus, in both
cases the structures are treated in a space group of lower symmetry.
Execute
script: runpp s/a... subst coord ffname
Input
files:
in.tp (unit 20), molecular
topology, linked to upack/data/subst/subst.tp$
in.spg (unit 11), space group, linked to upack/tops/all.spg
in.ff (unit 12), force field, linked to upack/tops/ffname.ff
in.pp (unit 13), code-directed input, linked to subst.pp#
in.cor (unit 29), one or more sets of coordinates, linked to coord
mol#.def (unit 48,
optional): a file defining additional charge sites on virtual atoms.
chadd.xyz (unit 48, optional): a file containing charges to
supersede the values from the topology file, and possibly also defining the
additional charges on virtual atoms.
Output
files:
out.pri (unit 16), general results, linked to prep.pr#
For every structure (identified with a four-digit
number #) there is the possibility for coordinate output in four formats (unit
17), linked to #.spf, #.xyz, #.mdi, #.cif, #.cssr.
For the grid search procedure (cf. pack1) the following two
files are produced:
out.exp (unit 18), summary of experimental data, linked to prep.exp.
out.tgt (unit 19), same data, but after energy minimization. For
the experimental structure this is a target function to be retrieved
"exactly", linked to prep.tg#.
The input data
can be manipulated by cards in file in.pp:
spgr naamsp {0}
If this card is absent or if naamsp = 0 the calculation is done for a free molecule. However,
this information can be overridden by space group information on the coordinate
file, which may be different for each subsequent structure.
inner inner{2}
intra intra
{1}
intra = 0/1:
disable/enable the calculation of intramolecular nonbonded interactions
flex iflex {1}
iflex = 0: rigid molecules, only intermolecular nonbonded
interactions. This option implies intra = 0.
iflex = 1: fully flexible molecules
iflex = 2: rigid molecules with rotatable OH groups, only
nonbonded interactions. The option naamsp = 0 is incompatible
with iflex = 2.
coul icoul {1}
icoul = 0: no Coulomb interactions are calculated.
icoul = 1: normal calculation of Coulomb interactions.
icoul = 2: the topology charges are replaced by the
charges from the CSSR file.
icoul = 3: the topology charges are replaced by the
charges from the file charges.cssr.
icoul = 4: the topology charges are replaced by the
charges from the file charges.xyz.
chadd ichad {0}
ichad = 0: charges from topology file
ichad = 1: charges from file chadd.xyz, but
only for real atoms
ichad = 2: charges from file chadd.xyz, also for
virtual atoms.
pres pres {0}
The pressure in atmospheres.
celopt icopt {1}
Cell axes and angles are kept fixed if icopt = 0.
expand expand {0} [nx ny
nz] {1 1 1}
If this code word is present, the coordinates are
expanded to P1. Random position disturbances (with maximum expand) can be added to
facilitate the minimization leaving a saddle point; a reasonable value might be
0.0005. Optionally, the structure is further expanded to nx, ny, nz unit cells in the a, b, c directions, respectively.
sd ncysd tolsd stpsd nresd shmsd {100 0.01 0.0001 5
0.02}
cg ncycg tolcg stpcg
depcg shmcg {5000 0.0005 0.005 0.3 0.02}
print iprint {0} [ipr1 ipr2 ipr3 ipr4
ipr5 {identical
to first parameter if unspecified}]
Minimal output on the screen is given for iprint =
ipr1:
reading of the coordinate file
ipr2:
calculation of bonded energies
ipr3:
calculation of nonbonded energies and forces
ipr4:
energy minimization
ipr5:
lattice vibrations
test dtest {0}
If dtest > 0, the analytical energy derivatives are compared
with numerically calculated values using parameter shifts dtest (dtest = 0.00001 is usually a good value).
conn icn {0 0 0….}
If the subroutine leesx fails, it
calls the subroutine connect to try and repair the situation. The geometry is compared
with the topology in order to assign the correct atom numbers to the
coordinates. There may be more solutions; usually they are identical but
in any case the program must be guided to select the correct solution number icn. It will ask you to enter that number manually. If you
know beforehand which one to choose, you may set conn with the desired
solution number(s) icn for each separate molecule.
reset
If this card is present, the C-H and O-H distances
are reset to values from the topology file.
cutoff cutoff
[alphac alpha6 hmaxc hmax6 tole tolk]
epsil epsil
Overrides the dielectric constant value given in the
force field file.
pol npp [poltol schaal gamma nnn mmm nte nttt]
roth dihin (1...Nh)
Reset dihedrals for hydroxyl groups to dihin (note: dihedral i
corresponds to ω(Nz+i))
Vn Vn (1...Nh) in kcal/mol {0.3...0.3}
ntal ntal (1...Nh) {3...3}
The latter two cards may be used if iflex = 2
stc stcin (1...6) {1 1 1 1 100 1}
Steepest descents weights for cell axes, cell axis
projections, centers of gravity, Euler angles, hydroxyl torsional angles, and
atomic coordinates, respectively.
files ifils
ifilm ifild ifilc {1 1 1 2}
molc irfat1 irfat2 {0 0}
Transforms atoms from irfat2 (molecule2) upwards to another equivalent position, such
as to make the distance between these atoms and irfat1 (in molecule 1) a minimum.
For a
free molecule only:
tors [dihin Vimp] (1...Nz+Nh)
{0 10000 ... 0 10000}
Target values for dihedrals. These values will be
kept (almost) constant during energy minimization by means of temporary
improper dihedral potentials Vimp (1...Nz+Nh) centered at dihin (1...Nz+Nh). If the chosen
dihedrals cause an internal collision, Vimp will have to be drastically reduced.
scan iscan
dihin(iscan) dscan escan
This card is only operative if tors is present. The
dihedral iscan is scanned from the
given value dihin(iscan) (which supersedes the corresponding entry from tors) in steps of dscan up to and including escan. The other dihedrals
wil be optimized during energy minimization.
knot
This codeword enables the search for knotted
structures.
Variant
for a free molecule with unknown coordinates
This is useful
to prepare starting geometries for the search procedure.
Execute script: runpp
s/a/... subst none ffname
The first call produces a
file cart.out. Copy
this file to cart.in and edit. It contains
indices of dihedral angles that have to entered manually (degrees). To minimize
the number of dihedrals to be assigned, the program omits hydrogen atoms in a
first try; if no chain of four atoms can be found, it tries again with hydrogen
atoms. If cart.in is present, the same
execute script will do a regular calculation. Check
the resulting geometry, especially the dihedral angles. Repeat the procedure
for every desired conformation, and rename the output file(s) 0000.mdi or 0000.spf to cor.#, the coordinate
file(s) to be used in the crystal structure generation.
If
lattice vibration calculations are desired:
latvib [nq ...] {0}
If this keyword is present, lattice vibration
calculations are performed. To this end double precision is required; it is
advisable to do energy minimization first. The calculations are first done for q
= 0, and subsequently for nq vectors in q-space. Possible modes of input:
latvib nq
Calculations are done for nq randomly chosen q-vectors.
latvib n1 n2 n3
Calculations are done for nq=n1 × n2 × n3 q-vectors, filling a regular grid in q-space.
latvib f1 f2 f3 nq
Calculations are done for the vectors defined by f1, f2, f3 divided in nq equal parts.
vibamp
If this card is present, eigenfunctions are
calculated in order to find the vibrational amplitudes.
temp vtemp(1....) {298, undefined ...}
The thermodynamical quantities (and the vibrational
amplitudes, if requested) are calculated at temperatures vtemp(1), vtemp(2), vtemp(3), ...
cutlv cutlv tollv {1.5, 0.0}
There is no Ewald summation possible, so one must be
able to choose a larger cutoff (cutlv) than in
the other calculations. Matrix elements smaller than tollv are
disregarded. Note that here the use of charge groups with appreciable net
charge may lead to inaccuracies.
celopt icopt dbox(1....) {1 0.01}
if icopt = 0, the cell axes and angles are kept fixed (but the cell
contents are optimized following sd and cg)
if icopt = 1, the cell axes and angles are optimized as usual
if icopt = 2, the cell axes and
angles are optimized followed by a search for the global minimum in the free energy.
This is done by a grid search of 3 points along each axis, separated by dbox(1). For determination
of the free energy lowering, this is usually sufficient. For thermal expansion
more iterations are needed, and to this end more entries dbox(2...) can be given. In
that case the search is iterated once for every entry, around the previous
minimum using the corresponding new grid separation. Since the minimum depends
on temperature, this is only done for the first temperature vtemp(1).
boxfix box(1...6)
This card starts the grid search for the global free
energy minimum directly at the given box parameters ax, by,
cz, bx, cx,
cy, again for the first temperature only.
This program
performs a molecular dynamics simulation for one structure. The coordinates of
that structure are given in file in.xvi which should be in mdi format (the coordinates
may be followed by the three velocities). After the simulation the output file out.xvo can be used as input
file for a continuation run.
Execute
script: runmd s/a/... subst coord ffname
Input
files:
in.tp (unit 20), molecular
topology, linked to upack/data/subst/subst.tp#.
in.spg (unit 11), space group, linked to upack/tops/all.spg
in.ff (unit 12), force field, linked to upack/tops/ffname.ff
in.md (unit 13),
code-directed input, linked to subst.md
in.xvi (unit 21), coordinates, linked to coord
Output
files:
out.pri (unit 16), general results, linked to md.pr#
out.en (unit 15), energy snapshots
out.co (unit 17),
coordinate snapshots
out.ve (unit 18),
velocity snapshots
out.xvo (unit 19), final coordinates and velocities
out.ave (unit 14), average results in MDI format
The input
specifications are given in file in.md, using the same code words as under the option mdrun
in program pack3. At least mdrun nstlim must be specified.
Additional code words are now:
spgr ispgr {0}
The number (not the name!) of the space group
ngr ngr {1}
The number of independent groups of molecules in the
unit cell
expand [nx ny
nz] {1 1 1}
If this card is present, the structure is expanded to
P1. Optionally, the structure is further expanded to nx, ny, nz unit cells in the a, b, c directions, respectively.
mdskip ig {0}
The number of runs to be skipped before block
averages of the remaining ones are taken.
print iprint {0}
References
1. D.A. Bardwell et al, Acta Cryst. B67 (2011), 535-551; A. M. Reilly et al, Acta Cryst. B72 (2016), 439-459.
2. B. P. van Eijck, W. T. M. Mooij
and J. Kroon, Acta Cryst.
B51 (1995) 99-103.
3. B. P. van Eijck and J.
Kroon, Acta Cryst. B56 (2000) 535-542.
4. B. P. van Eijck, J. Comput.
Chem. 23 (2002) 456-462.
5. A. L. Spek, Acta Cryst.
A46 (1990) C34.
6. W. F. van Gunsteren and
H. J. C. Berendsen, GROMOS, Groningen molecular simulation package, University of Groningen, 1987.
7. B. P. van Eijck and J.
Kroon, J. Comput. Chem. 20
(1999) 799-812.
8. B. P. van Eijck, J. Comput.
Chem. 22
(2001) 816-826.
9. W. Damm, A. Frontera, J.
Tirado-Rives and W. L. Jorgensen, J. Comput. Chem. 16
(1997) 1955-1970.
10. D. E. Williams and T. L. Starr, Comput. Chem. 1 (1977) 173-177.
11. B. P. van Eijck, A. L. Spek, W. T. M.
Mooij and J. Kroon, Acta Cryst.
B54 (1998)
291-299.
12. D. N. Bernardo, Y. Ding, K. Krogh-Jespersen and
R. M. Levy, J. Phys. Chem. 98 (1994) 4180-4187.
13. B. P. van Eijck and J.
Kroon, J. Phys. Chem. B101
(1997) 1096-1100.
14. W. F. van Gunsteren and
H. J. C. Berendsen, Angew. Chem. Int. Ed. Engl. 29 (1990)
992-1023.