Crystallographic Methods
Research Goals
In our research, we continuously strive to improve crystallographic (refinement) methods to get the most out of our crystals.
With increasing automation of synchrotron beamlines, it is now possible to collect more and more datasets from one or many crystals of a protein. We are currently developing methods to take advantage of these repeated observations, improve our models and help to identify new structural features.
Updates coming soon -- watch this space. In the meantime, you can take a look at some of Nick's previous multi-dataset work.
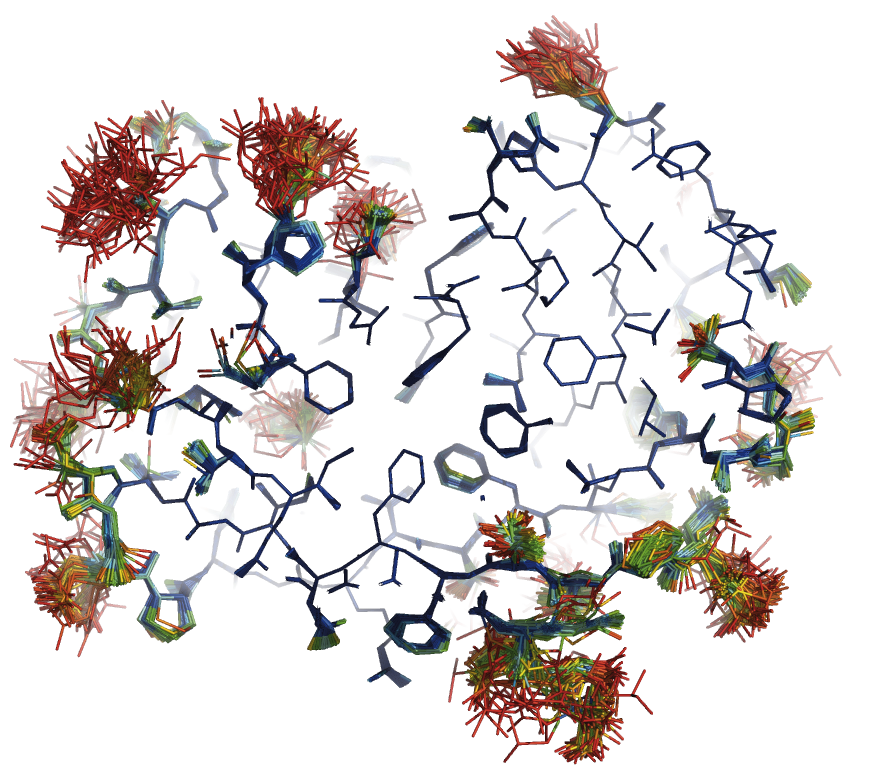
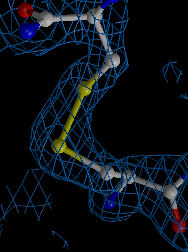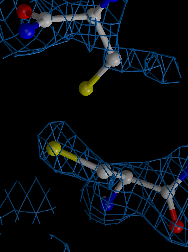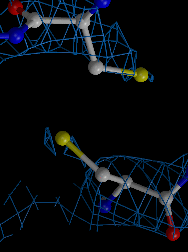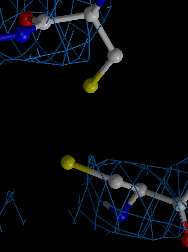
We developed a method for determining biomacromolcular structures from X-ray diffraction data that accounts for the flexibility or dynamics of the molecules (Burnley et al. eLife 2012). The method combines Molecular Dynamics simulations with experimental diffraction data evolving the traditional single and static structure into a dynamic ensemble model containing multiple structures. This development builds on the orginal idea of "time-averaging" in crystallography (Gros et al. Science 1990) using modern tools (such as maximum likelihood refinement and TLS), now producing ensemble models that show improved Rfree values compared to traditional single structure refinements.
phenix.ensemble_refinement is available as part of the PHENIX software suite.

We developed a N-particle method called Conditional Optimization that allows the incorporation of extensive prior geometrical data into protein-structure determination before a reliable model can be constructed. We tested conditional optimization for refinement, automated model building and ab initio phasing. Refinement revealed an enhanced radius of convergence. Automated model building yielded models of comparable phase quality to those obtained from the commonly used programs ARP/wARP and RESOLVE. For ab initio phasing we demonstrated that the method works successfully on theoretical data (see movie that shows an optimization run that started from a random distribution followed by energy minimization and dynamics optimization). Preliminary experiments on phasing using real data indicate that the method works but is currently too slow for practical use.